
|
|
| Home | Research | Publications | People | Joining the Group | Teaching |
|
The topochemical deintercalation of oxide ions from extended transition metal oxide lattices changes both the oxidation state of transition metal centres, and their local coordination environments two features which have a decisive influence on the electronic and magnetic behaviour of phases. We have demonstrated that binary metal hydrides (NaH, LiH, CaH2) can be used to deintercalate oxide ions from complex transition metal oxide phases to yield highly metastable phases which contain transition-metal centres in highly unusual oxidation states and/or coordination environments. |
|
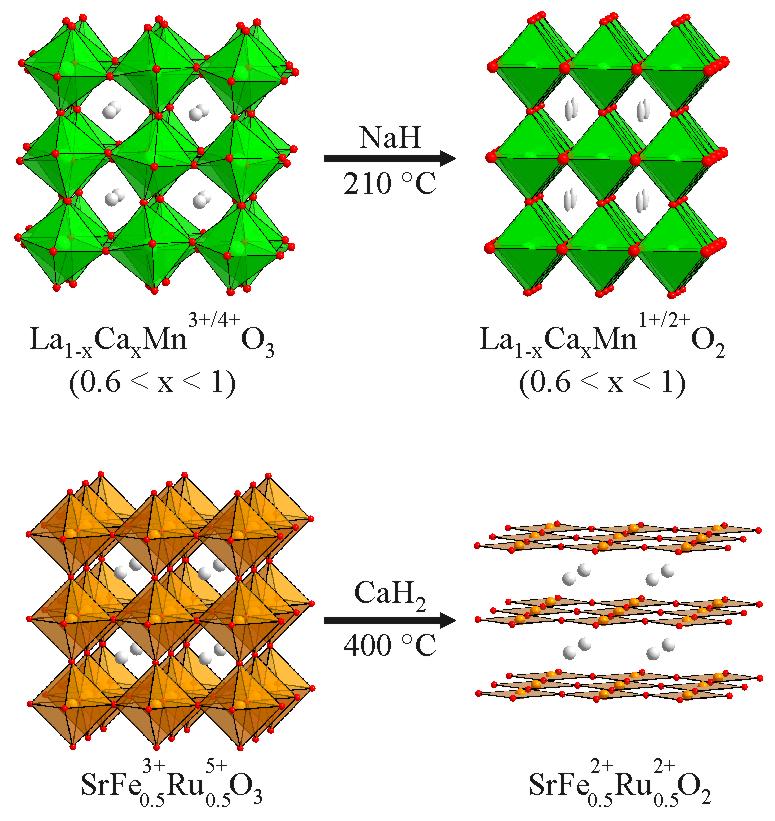
Unusual Transition-Metal Oxidation States We have demonstrated that binary metal hydrides (NaH, LiH, CaH2) can be used to deintercalate oxide ions from complex transition metal oxide phases to yield highly metastable phases which contain transition-metal centres in highly unusual oxidation states. For example, by reacting the Mn3+/4+ perovskite phase La1-xCaxMnO3 (0.6 < x < 1) with NaH at 210 ºC, oxygen can deintercalated from the structure to form La1-xCaxMnO2 - a material which retains the cation lattice of the parent phase but has a lower oxygen stoichiometry. During the reaction the oxide ion lattice reorganises itself to retain the octahedral coordination of the manganese cations to yield a material which can be thought of as a cation-ordered variant of the rock-salt structure. The decrease in the oxygen stoichiometry reduces the manganese cations, so they have an average oxidation state less than Mn2+. This is the first reported example of Mn1+ in an extended oxide phase. In contrast the reaction of SrFe0.5Ru0.5O3 with CaH2 at 400 ºC yields SrFe0.5Ru0.5O2, a phase in which both iron and ruthenium occupy square-planar coordination sites and have both been reduced to the divalent oxidation state. This is the first example of Ru2+ in an extended oxide. Magnetisation measurements and DFT calculations indicate the d6 Fe2+ centres adopt a high-spin S = 2 configuration, while in contrast the d6 Ru2+ centres adopt an intermediate spin S = 1 configuration, leading to net spin-glass behaviour. Journal of the American Chemical Society
133 (2011) 18397. |
 |
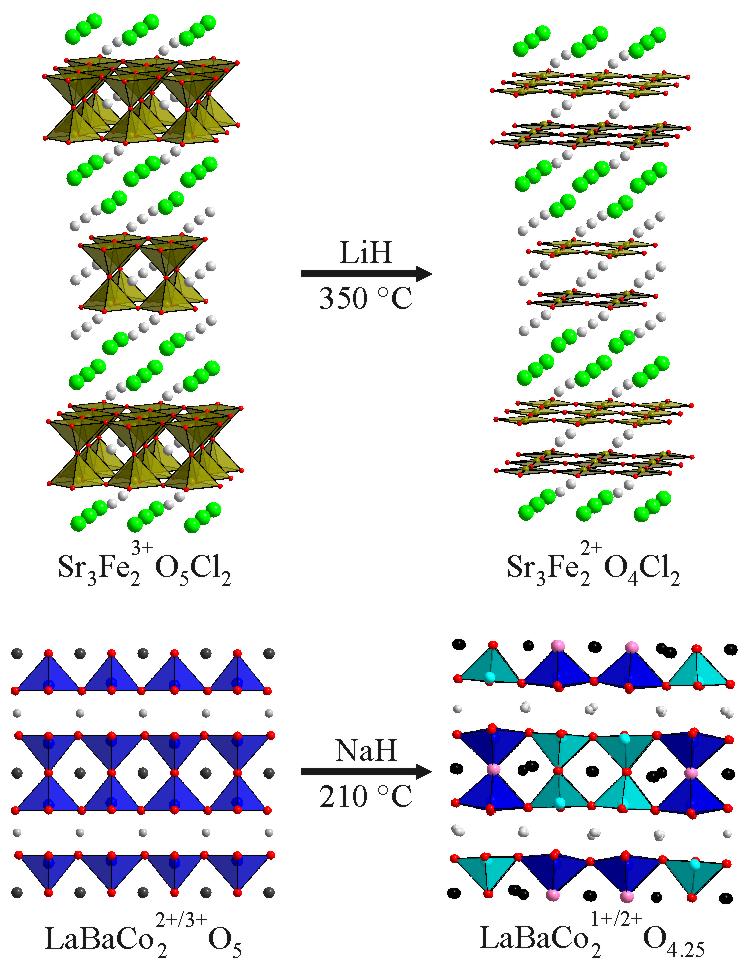 |
Unusual Transition-Metal Coordination Geometries The local coordination geometry of ligands around transition metal cations defines the relative energies of the metal d-orbitals, and thus has a strong influence on the electronic structure and behaviour of extended metal oxides. The topochemical nature of low-temperature anion deintercalation reactions means that phases prepared in this way are not restricted to forming only the most energetically favourable coordination polyhedra (e.g. octahedra and tetrahedra) but can instead form materials with more exotic point symmetries at metal centres. For example, reaction of the oxy-chloride phase Sr3Fe2O5Cl2 with LiH yields Sr3Fe2O4Cl2, a material containing sheets of apex-sharing FeO4 square planes. Further investigation reveals that the local coordination of the iron centres in Sr3Fe2O4Cl2 is directed by the surrounding lattice, so that by replacing the iron with cobalt, the isostructural phase Sr3Co2O4Cl2, containing square-planar Co2+, can be prepared. The square-planar Co2+ centres adopt an orbitally degenerate electronic configuration so that the system undergoes a structural symmetry breaking transition below 200K, due to a cooperative Jahn-Teller distortion. The influence of spectator cations can also be seen during the reduction of REBaCo2O5 phases. Reaction of YBaCo2O5 with NaH yields YBaCo2O4.5, a Co2+ phase which contains CoO4 tetrahedra and CoO5 square-based pyramids. In contrast the reduction of the isostructural phase LaBaCo2O5 yields LaBaCo2O4.25 a phase that not only contains Co2+ centres in CoO4 tetrahedra and CoO5 square-based pyramids, but also Co1+ centres in square-planar coordination sites.This demonstrates that the identity of spectator cations can influence both the local metal coordination geometries and oxygen stoichiometry of product phases. Inorganic Chemistry 49 (2010) 11062. |