 Image credit: Carlos Reding
Image credit: Carlos Reding
Image credit: Carlos Reding
Image credit: Carlos Reding
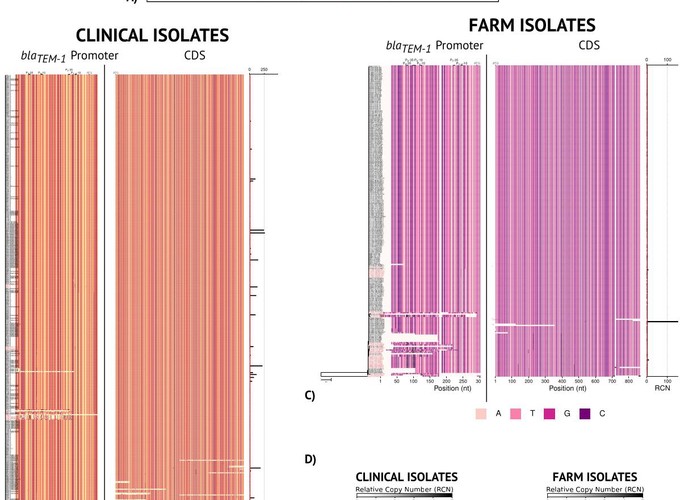
Increasing evidence suggests that microbial species have a strong within species genetic heterogeneity. This can be problematic for the analysis of prokaryote genomes, which commonly relies on a reference genome to guide the assembly process: any difference between reference and sample genomes can introduce errors in the detection of small insertions, deletions, structural variations and even point mutations. This phenomenon jeopardises the genomic surveillance of antibiotic-resistant bacteria, triggering even a reproducibility crisis. Here we present Hound, an analysis pipeline that integrates publicly available tools to locally assemble prokaryote genomes de novo, detect genes by similarity, and report the mutations found. Three features are exclusive to Hound: It reports relative gene copy number, retrieves sequences upstream the start codon to detect mutations in promoter regions, and, importantly, can merge contigs based on a user-given query sequence to reconstruct genes that are fragmented by the assembler. To demonstrate Hound, we screened through 5,032 bacterial whole-genome sequences isolated from farm animals and clinical patients using the amino acid sequence of blaTEM-1, to predict resistance to amoxicillin-clavulanate. We believe this tool can facilitate the analysis of prokaryote species that currently lack a reference genome, and can be scaled up to build automated diagnostic systems.