First Published in:
Diagnostic and Therapeutic Antibodies
ed AJT George and CE Urch
Methods in Molecular Medicine vol 40, pp 243-266, pub Humana Press, Totowa, NJ
Geoff Hale, 1998
Humana Press, 2000
Posted on the internet with the permission of the copyright holders.
In 1890 it was shown that resistance to diphtheria toxin could be transferred from one animal to another by transfer of serum [1]. From this discovery, passive antibody therapy was developed as an effective treatment for infectious diseases and for neutralization of toxins, and continues to be used to this day. Meanwhile, there have been continued efforts to use antibodies for cancer therapy, starting with the pioneering work of Hericourt and Richet in 1895 [2] which was the forerunner of the "magic bullet" concept. However, all of the early work on tumour therapy led ultimately to disappointment [3]. The problems were readily acknowledged, ie lack of specificity and reproducibility, lack of purity and the xenogeneic immune response. Developments over the past 20 years, as described throughout this book, have effectively overcome all of these technical problems, often in very ingenious ways. The difficulty we have now is different. There are just too many potential new antibody based treatments for them all to be properly evaluated in the clinic. Many will still fail because of factors which are hard to predict from experiments: unexpected toxicity, biological heterogeneity of the target disease or lack of access to the appropriate tissue.
The unfortunate reality is that clinical development of new drugs is almost entirely in the hands of the pharmaceutical industry, where many factors other than strictly scientific/clinical ones influence the decision to develop a particular product. These include the availability of patent protection, the potential size of the market, the impact of new drug regulations and the current mood of investors. Profitability is the over-riding concern and is an essential ingredient of success in our society. However, we do not think it is enough for academic scientists to patent a new technology, license it on to industry and sit back with large up-front payments. Physicians and their patients are cut out of the decision process and become essentially a commodity to be bought or discarded during the clinical trials. A better way is for scientists, physicians, patients, industry and government to work together, each recognising the different priorities of the other, but each contributing to the decision process. Biological therapies will be costly and they do carry risks of introducing dangerous new diseases. We all have an interest in their effective application.
Here we will tell the story of CAMPATH-1, just one of many hundreds of monoclonal antibodies developed for therapy. It illustrates several aspects of practical development which might apply to other projects and perhaps the reader will be able to avoid the pitfalls which we encountered. CAMPATH-1 was one of the first therapeutic monoclonals to be assigned for commercial development and it was licensed to one of the best equipped UK Biotech companies of the time. However, were it not for the dedication of teams of clinicians worldwide who continued to pursue independent trials with our academic group, we are convinced that it would by now have disappeared without trace.
The original impetus for development of CAMPATH-1 came from bone marrow transplantation. Since monoclonal antibody technology was invented by immunologists, it was not surprising that they looked to immunological diseases for some of the first applications. Graft-versus-host disease (GvHD) was a good target, since it was known to be caused by T cells which inevitably contaminated donor bone marrow. If the T cells were removed before transplantation, GvHD and its potentially lethal effects could be eliminated. Monoclonal antibodies specific for T cell antigens were discovered by several groups in the late 1970‘s and early 1980‘s. Antibodies alone are not usually toxic to cells and they needed to be used with a second reagent to kill or separate the unwanted T cells [4,5]. An obvious choice was complement, well known as the natural effector arm of humoral immunity. However, unlike polyclonal antisera, the majority of monoclonal antibodies were poor at activating complement and it was particularly difficult to kill human cells with human complement. Now we know that there are several reasons: the choice of antibody isotype, the limited density of most cell surface antigens, the propensity of some antigens to patch and cap and the presence of cell-surface inhibitors of complement, like decay accelerating factor and CD59. At the time, these problems were partly overcome using heterologous serum (eg from rabbit) as a source of the complement because the human cell surface inhibitors failed to inactivate the heterologous complement cascade. However, this introduced an unwanted source of heterogeneity and it was difficult to procure reliable and non-toxic batches of rabbit serum. Accordingly our group continued to search for an antibody which would activate human complement, bind to T cells and spare stem cells. The goal was "operational" specificity, not necessarily lineage restricted; a novel concept at the time.
In 1979, Herman immunised a rat with human lymphocytes and carried out a fusion which was to yield an amazing diversity of antibodies. The choice of rats rather than mice for the fusion was made for a number of technical reasons [6], but it turned out to be truly serendipitous. Antibodies were identified by a range of assays carried out by many team members, and many of the reagents discovered have been used in basic research and human therapy. Several antibodies were found which lysed T cells with human complement. At this point Geoff joined the lab, a young protein chemist given the task of characterising the structure of these new antigens - a task in which he was singularly unsuccessful for many years. It turned out that all of the lytic antibodies recognised the same antigen, which we now know as CD52. The majority of other antibodies from the fusion were rat IgG2a or IgG2b, but the CD52 antibodies were mostly IgM, with one IgG2c and one IgG2a (YTH34.5). There is a strong isotype bias in the rat antibody response; IgG2a and IgG2b are commonly made against surface protein antigens, IgG1 against soluble protein antigens and IgM or IgG2c against T cell independent antigens (which we originally equated with carbohydrates - incorrectly in this case). Since then, other IgM and IgG2c CD52 antibodies have been obtained but the YTH34.5 clone is still unique. It later proved to be absolutely crucial for clinical success. However, one of the IgM antibodies (clone YTH66.9, CAMPATH-1M) was originally chosen because it activated complement more efficiently and gave virtually complete elimination of T cells in vitro.
At that time our lab was fortunate to have two postdocs from the leading centres of progenitor cell biology: Sue Watt from Don Metcalf‘s lab in Melbourne and Trang Hoang from Norman Iscove‘s lab in Lausanne. Using their respective culture systems, we showed that the CAMPATH-1 antigen was not expressed on bone marrow colony-forming cells [7,8]. CAMPATH-1 antibodies cross-reacted with lymphocytes from non-human primates and safety studies were initiated in baboons and cynomolgus monkeys [8,9]. It was not until the first animals were injected that we realised that the antigen was also expressed on the red cells. There was haemagglutination and haematuria but animals remained well! Further studies showed that the red cell antigen is polymorphic, expressed in only some monkeys whereas it is always found on lymphocytes; a fortunate situation which allowed the safety studies to proceed but is still not explained. Screening of all available human blood groups by our colleagues in the transfusion centre, showed that the CD52 antigen is never expressed on human red cells. At the same time David Swirsky working with Frank Hayhoe in the Haematology Department next door to us, treated a patient suffering from end stage non-Hodgkin‘s lymphoma [9]. The clinical effect was minimal, just like all the other monoclonal antibodies being tested at that time. There was temporary clearance of tumour cells from blood, accompanied by consumption of complement, but 24h after each dose the cells bounced back. Nevertheless, the treatment was well tolerated and we felt that the stage was set for the first trial in bone marrow transplantation. With high hopes, we submitted our first paper describing this new specificity and its potential clinical application [7]. We were greatly disappointed when it was comprehensively rejected, first by the Journal of Experimental Medicine and then by Blood. It was only by a personal plea from Don Metcalf that the editors of Blood were persuaded to change their mind.
Our work had been funded by the UK Medical Research Council (MRC). In those days, Universities had no organisation for commercial exploitation of inventions but the MRC had a commitment to pass any discoveries by their employees to the National Research and Development Council (NRDC) - a government organisation devoted to technical transfer. NRDC is famous for its decision when offered the pioneering discovery of Kohler and Milstein: "It is certainly difficult for us to identify any immediate practical applications which could be pursued as a commercial venture". This probably did more than anything else to facilitate the widespread use of monoclonal antibodies, but no doubt the MRC regrets the untold millions of lost royalties! Although we were not bound by the rules for MRC employees, we felt that it was right to offer CAMPATH-1 to NRDC (now renamed "British Technology Group", BTG) before the work was published. In a recapitulation of the earlier decision, BTG decided that there was no need to patent the CAMPATH-1 specificity or its proposed application. However, they did acquire all of the rights to the CAMPATH-1 cell lines. We were näive about legal agreements and did not appreciate the gulf between "assignment" and "licence". We simply imagined that a government organisation set up to exploit British discoveries was the obvious way forward. There was no alternative. Luckily we did obtain a clause allowing us to continue our own academic and clinical research with the cell lines which now belonged to BTG.
The first bone marrow transplant using CAMPATH-1M for T cell depletion was carried out in 1982 at the Hammersmith hospital by Jill Hows and Ted Gordon-Smith. The outcome was a disaster from the point of view of our research plans but the antibody treatment probably saved the life of the patient! The patient suffered from severe aplastic anaemia - failure of the bone marrow, which could only be cured by a bone marrow transplant. This patient had no suitable HLA- matched sibling donor, so it was decided to try a transplant from an unrelated donor. Such transplants were very experimental at the time and it was realised that there would be a high risk of GvHD. Initial optimism on seeing the first signs of marrow recovery turned to a disappointment mixed with relief when it was found that the blood cells were solely of recipient type. Remarkably, the patient‘s own bone marrow had started to work again and the lady remains alive and well to this present day. Although the preclinical work had convinced us that stem cells should be spared, it was hard to explain the outcome and the physicians, not unreasonably, called a halt to the clinical programme. Now we realise that T-cell depletion increases the risk of graft rejection, which was already high in an unrelated transplant for a multiply-transfused patient suffering from anaemia.
A chance meeting between Steve Cobbold and Shimon Slavin from the Hadassah hospital in Jerusalem, with some vital encouragement from Cesar Milstein, led to the next step. Shimon had been conducting experimental and clinical marrow transplantation for some years and realised that T cell depletion was the most promising method to prevent GvHD, which was still the major clinical problem. He took the chance to evaluate CAMPATH-1M in a small series of patients transplanted from HLA- matched siblings. Although they all had poor prognosis leukaemia, at least the risks of immunological complications were not so extreme. The successful results of this pilot study, published in the Lancet in 1984 [10], were soon emulated by several other transplant groups in Europe, including teams in Ulm, Germany [11] and again at the Hammersmith hospital in London [12]. This led to the formation of the CAMPATH users group, a convivial and informal association of transplant centres worldwide, which was the prototype for many other clinical collaborations over the following 15 years.
CAMPATH-1M was remarkably effective at preventing GvHD, reducing the incidence from about 40% to 10% or less but a new problem was seen - graft rejection - which now affected 15-20% of the patients and led to a number of fatalities [13]. This urgently needed to be tackled if there was to be any further progress. Since 1979, in parallel with the studies on antibodies against human cells, Steve Cobbold had been working to develop analogous anti-mouse antibodies to study transplantation in the laboratory. He found that certain rat monoclonal antibodies, notably of the IgG2b isotype, could deplete mouse T cells very effectively [14]. There was no homologue of CAMPATH-1 but rat IgG2b anti-Thy-1 or anti-CD4 plus anti-CD8 could prevent GvHD, and with the same outcome as in humans - ie graft rejection. Reasoning that graft rejection was due to residual host T cells which had been spared by the standard conditioning regimen, Steve pre-treated the recipients with the same cocktail of depleting antibodies. This time the mice survived with full engraftment and no GvHD [15]. Obviously we wanted an antibody which could produce a similar depletion in vivo in humans. The likely mechanism for cell depletion was ADCC, mediated by antibody binding to Fc receptors on NK cells or macrophages. By screening a large panel of rat monoclonals we found that the only isotype which gave effective ADCC with human cells was rat IgG2b [16], but we had no CD52 antibodies of this isotype. However, it was known that hybrid myeloma cells could occasionally switch isotypes in vitro, in a parody of the normal physiological progress of class-switching. At the time Marianne Bruggeman had joined our lab and was starting to map the rat immunoglobulin gene locus [17]. She showed that the IgG2b constant region was downstream of IgG2a, so there was a chance that we could select a suitable mutant of YTH34.5. Rat hybrid myelomas are more stable than mouse and after screening about 20 million clones, Geoff found 34 which had switched to IgG2b [18]. Many of them showed a strong tendency to switch back again, but ultimately a stable clone was isolated. This was the antibody we named CAMPATH-1G.
A clinical opportunity to test the new antibody was soon presented. A patient suffering from CLL in prolymphocytic transformation had rapidly progressive disease which had responded only partially to chemotherapy [19]. His tumour cells were sensitive to CAMPATH-1G in vitro and so Martin Dyer and Frank Hayhoe decided to proceed with therapy. The initial results were beyond our most optimistic expectations. Two days of treatment with CAMPATH-1M gave only a very transient response as before, but CAMPATH1G immediately started to clear tumour cells. After 10 days of treatment, the blood and bone marrow were completely cleared and by normal criteria the patient was in complete remission. Many sincere prayers had been offered for this man and it did cross our minds that divine intervention might have over-ruled our efforts! Sadly this was not to be the case; just a few days later, signs of florid CNS disease became evident, and large numbers of tumour cells were discovered in the cerebrospinal fluid. We knew that the antibody was unlikely to cross the blood-brain barrier and so in a last effort, CAMPATH-1G was infused direct into the cerebrospinal fluid. This was well tolerated, but unfortunately had no impact on the tumour cells; the next day there were just as many and they were all uniformly coated with antibody. The patient did not respond to other therapies and died of progressive disease a few weeks later. Despite the tragic outcome, this single clinical experiment was very informative, showing the vital role of effector cells (absent from the CNS) and gave us hope that other patients could be successfully treated. It was a crucial turning point in the development of CAMPATH-1.
The second patient was another uniquely informative case [19]. She was a lady suffering from CLL who in 1985 had been treated with both YTH34.5 (the original IgG2a) and CAMPATH-1M. Both had only a very transient effect, just like other patients who had been treated with the IgM antibody alone. For two years her disease had been held in check by chemotherapy, but now it had transformed and started to rapidly progress. The effects of CAMPATH-1G were again dramatic, resulting in a clearance of the majority of tumour cells from blood, but only a modest reduction of tumour infiltration in the bone marrow (from 97% to 87%). After a few weeks, tumour cells started to reappear in the blood, albeit at a much lower level than before. The patient had made an immune response against the rat monoclonal antibody which neutralised the CAMPATH-1G and limited its effectiveness. We knew that a more nearly human antibody was needed and by good fortune the technology to achieve this was being developed by Michael Neuberger and Greg Winter just across the road in the MRC Laboratory of Molecular Biology [20,21].
But first, we should tell some of the other developments which had been occurring. BTG had been set up with the aim of enabling UK companies to exploit inventions from British academia. They started negotiations with Celltech (now Lonza) in 1983 and continued for two years without reaching agreement. Then in 1985 CAMPATH-1M was licensed to Wellcome Biotech, a small subsidiary of the giant Wellcome Foundation. We were pleased because Wellcome had an important reputation in production of biologicals, being the first company to establish a process using a continuously cultured mammalian cell line (production of a-interferon). Clinical trial material was produced and used in a small number of bone marrow transplants, but the outcome was never evaluated. CAMPATH-1G had much more promise for widespread applications: treatment of leukaemia and lymphoma, as an immunosuppressive agent for organ transplantation [22] and perhaps autoimmune disease. Because the original YTH34.5 clone now belonged to BTG, we were obliged to hand over CAMPATH-1G as well and this in turn was licensed to Wellcome Biotech. For the second time they started the process of preparing material for clinical trials.
In 1986, Greg Winter and his team published the first humanised antibody created by genetically grafting the CDR regions from a mouse antibody [21]. The antibody was directed against a chemical hapten, just a model with no therapeutic application, and in fact only the heavy chain was humanised. The obvious next step was to humanise a therapeutic antibody and it seemed that CAMPATH-1G was an ideal choice. Lutz Riechmann, a molecular biologist in Greg Winter‘s lab and Mike Clark, a postdoc who had joined our lab, worked together to engineer a whole set of chimeric and humanised antibodies [23]. At the time, we did not know how efficient would be the association of CDR-grafted variable regions. Also, we could not be sure which human isotype would have the best effector functions, although model studies by Marianne Bruggeman, Mike Clark and Carol Bindon were showing IgG1 and IgG3 to be good candidates [24]. Therefore the project proceeded in painstaking steps, each intermediate construct being expressed and tested for activity. Ultimately a whole panel of chimeric and humanised antibodies were created of different IgG subclass which allowed Mike to confirm that IgG1 is the most potent for both activation of complement and ADCC. Subsequently many more variants have been created, including different IgG allotypes, null allotypes, domain shuffled, point mutants etc. and they have allowed an ever more detailed dissection of the interaction of human IgG with physiological effector mechanisms. These rules have guided the design of all subsequent therapeutic antibodies.
Our team was hugely excited by these results and we felt this was a watershed in the development of the long-awaited "magic bullet". BTG did not agree. They were concerned that CAMPATH-1H might compromise the investment already made in CAMPATH-1G and expressed their disapproval of the collaboration with Winter‘s group in no uncertain terms! The only thing to do was to hand over CAMPATH-1H to them, especially since this seemed to offer the best route to commercial development via a simple extension of the existing licence to Wellcome Biotech. We had developed good relationships with the Wellcome Biotech team and felt confident that the senior management had a big commitment to the project. The company abandoned work on CAMPATH-1G and started in earnest to develop CAMPATH-1H, which they expected could reach a much wider market and perhaps break through the "billion dollar threshold" which big pharma are seeking for. Over the next seven years, Wellcome were to devote maybe Ł50M to the development of CAMPATH-1H.
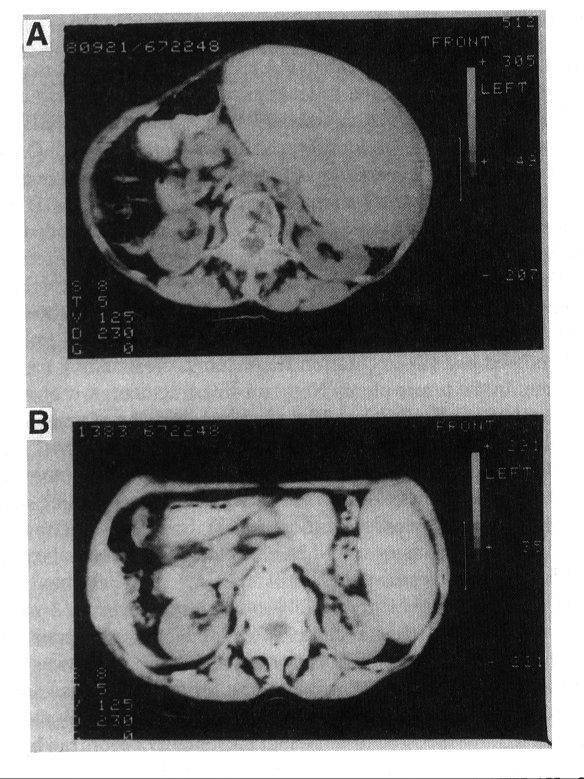
The first patient to be treated with CAMPATH-1H was a lady suffering from non-Hodgkins lymphoma in leukaemic phase [25]. She had a very high white cell count, bone marrow completely packed with tumour cells and gross splenomegaly. Six months previously, she had been treated with CAMPATH-1G which gave a partial response but had been discontinued because of an allergic type of reaction. Only a small amount of CAMPATH-1H was available and it was administed in small doses (1-20 mg) over a period of 30 days (total 120 mg). Even to an untrained observer the response seems quite remarkable (Fig 1).
Figure 1. Treatment of non-Hodgkin‘s lymphoma with CAMPATH-1H.
Computed tomographic scans showing the extent of splenomegaly before (A) and after (B) treatment. (Reproduced with permission from ref 25.)
The spleen size was reduced from 4.5 kg to 0.6 kg. Tumour cells could no longer be found in the blood or bone marrow and they began to be replaced by normal blood elements. Analysis of DNA from a bone marrow aspirate before treatment showed just two restriction fragments corresponding to biallelic Ig rearrangement in the tumour clone. No germ-line fragments were visible at all. In marked contrast, a sample taken 28 days after the beginning of treatment showed only germ line genes and no sign of the Ig gene from the tumour (Fig 2).
Figure 2. Clearance of tumour cells demonstrated by Southern blotting to detect residual clonal lymphocytes following treatment with CAMPATH-1H.
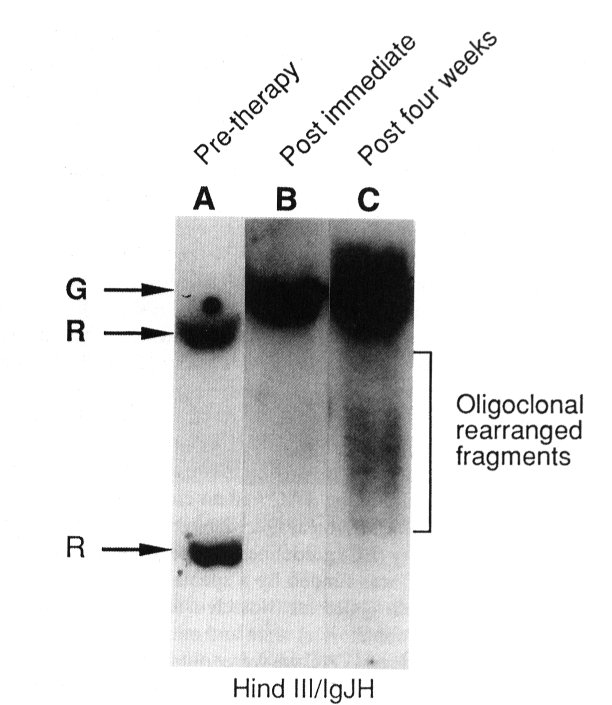
DNA was extracted from bone marrow mononuclear cells, digested with restriction endonuclease HindIII and probed with an immunoglobulin JH DNA probe. Lane A (before antibody therapy) shows biallelic IgJH rearrangement (R) with no detectable germline band (G). Lane B (directly after antibody therapy) shows germline fragment only. Lane C (4 weeks later) shows oligoclonal rearranged fragments, interpreted as representing re-emergence of normal B cell progenitors. The tumour clone is still not visible. (Reproduced with permission from Dyer et al, Leukemia and Lymphoma, 2, 179-193.)
This technique was sensitive to about 1 in 400 tumour cells. An even more encouraging observation was that a sample taken 11 weeks after the beginning of antibody treatment still showed none of the tumour-specific fragments, but now there were a spread of new fragments as would be expected for oligoclonal expansion of normal B cells. However, the patient still had a reservoir of tumour cells in the spleen and relapse was evident in the blood and bone marrow 3 months after the beginning of treatment. She received a second course of treatment with CAMPATH-1H with similar response and then the spleen was surgically removed (which would have been impossible originally because of its large size). Histological examination of the spleen showed very many macrophages which appeared to have ingested tumour cells; presumably opsonised by the monoclonal antibody. Fifteen months later the patient relapsed again and died shortly after. This lady had been unusual in that she had no bulky lymph nodes detectable by CT scanning. Previous trials with CAMPATH-1G in 18 patients with a variety of lymphoid malignancies had shown that tumour cells could generally be cleared from blood, frequently from bone marrow and spleen, but lymph nodes and extranodal masses were very rarely affected. We were therefore very interested to know whether the humanised antibody would be any better. The second patient to be treated with CAMPATH-1H was Ken Sander, a retired professor of electrical engineering. He had been newly diagnosed with stage IVA grade 1 non-Hodgkin‘s lymphoma, again with a high white cell count. Enlarged lymph nodes were visible by CT scan. Treatment with CAMPATH-1H induced a complete remission by clinical criteria, including regression of the lymph nodes. However, Bence-Jones protein (indicative of residual disease) was detectable within a few months and so he continued to be treated with conventional chemotherapy. During the coming years, Prof Sander and his family engaged in many fund-raising activities to help our research - their commitment motivated us enormously.
The original rat monoclonal antibodies had been prepared from ascitic fluid, crudely fractionated with ammonium sulphate. Quality control consisted of little more than measurement of binding activity and checks to ensure sterility and absence of pyrogens. In 1987 we had obtained a hollow-fibre cell culture device (Acusyst-Jr) and started to produce CAMPATH-1G in the laboratory. This was the only way to produce CAMPATH-1H if we wanted to exclude contamination with rat proteins. There were other good reasons to avoid in vivo production - an immune response might be made against the engineered cell line with its human genes, there were concerns about the unnecessary use of animals, and the need to maintain a colony free of potential pathogens would become very onerous. However, it was becoming obvious that in an ordinary research lab we could not carry out the large amount of cell culture, processing and quality control to make products for clinical trials. Quite aside from the scale and costs, the potential for cross-contamination was too great. Commercial production was not yet underway and there was a growing demand from our clinical colleagues to expand the scope of their trials. All of the production work was being done by one postdoctoral scientist, Jenny Phillips. In 1990, we set up a new group, the Therapeutic Antibody Centre (TAC), devoted entirely to antibody production for clinical research. We did not intend to engage in routine production or definitive clinical trials which are more properly the role of pharmaceutical industry, but we wanted to liberate clinical research from the bottleneck of pilot scale production. Initially the TAC was based in the Blood Transfusion Centre at Cambridge, as a fore-runner of what we believed to be a new and vital role for the Transfusion Service in the development of biological therapies [26]. From the beginning the TAC had an emphasis on quality and with the appointment of Patrick Harrison as QA Manager we strove to meet the emerging EC guidelines for control of manufacture of biological products. The TAC was funded by a special grant from the Medical Research Council together with a contribution from Wellcome Biotech in recognition of the growing collaboration between the commercial team and our research group. However, on the opening day of the new TAC we were told that Wellcome Biotech was to be wound up and reabsorbed into the parent company. We knew that they had suffered during a bruising patent battle with Genentech about tissue-plasminogen activator, and we were sad to hear this news which meant that several of the senior management would shortly be moving elsewhere. Although CAMPATH-1H continued to be developed, and we enjoyed a productive relationship with many of the Wellcome scientists, the ultimate decisions were removed to a level beyond our access, and clinical trial protocols were developed with a focus on large markets, starting with non-Hodgkin‘s lymphoma and subsequently rheumatoid arthritis. It was unfortunate that these turned out to be two of the less favourable indications.
A couple of months after the TAC was opened, Martin Lockwood approached us about Nicola Cole, a young lady who was suffering from the effects of a very severe auto-immune disease, an unusual type of vasculitis characterised by T cell infiltration of the blood vessels [27]. Although Martin has great experience with this disease and had tried various standard and experimental therapies, there seemed to be nothing that would work. To our delight, a short course of treatment with CAMPATH-1H had a swift effect and soon it was possible to withdraw the palliative intravenous morphine and after a little while she was able to return to normal activities for the first time in several years. Over the course of the next four years, she required further treatment with CAMPATH-1H on seven occasions. Each time the treatment resulted in a disease remission, but after three courses an anti-idiotype response was detected against the humanised antibody. Further courses were successfully given after temporarily removing the anti-id antibody by plasmapheresis, but eventually the titre became too high for this to be practicable. She was one of only a very few patients who have ever produced a strong anti-globulin response to CAMPATH-1H, but her antiserum has been of great value to us in defining the antigenic epitopes and guiding new developments designed to overcome this side effect. We learned a great deal from this single case, not least about the remarkable endurance and determination of patients who volunteer for clinical trials and put up with many interventions and investigations to provide data which will help others. It was a great pleasure for us when in 1995 Nicola came to open our new Therapeutic Antibody Centre in Oxford (Fig 3).
Figure 3. Nicola Cole and Geoff Hale at the opening of the Therapeutic Antibody Centre, Oxford, December 1995.

Since that first case, Martin Lockwood has gone on to treat more than 70 other patients, not only with the rare T cell forms of vasculitis, but also including more classical cases (eg Wegener‘s granulomatosis) where auto-antibodies are present [28]. Remissions were induced in virtually all patients lasting from 2 months to more than 2 years, allowing a substantial reduction in the normal steroid therapy, with obvious clinical benefits (Fig 4). As a general rule, patients who relapsed have been successfully retreated and there have been few other examples of a significant anti-globulin response.
Figure 4. Treatment of Wegener‘s granulomatosis with CAMPATH-1H.
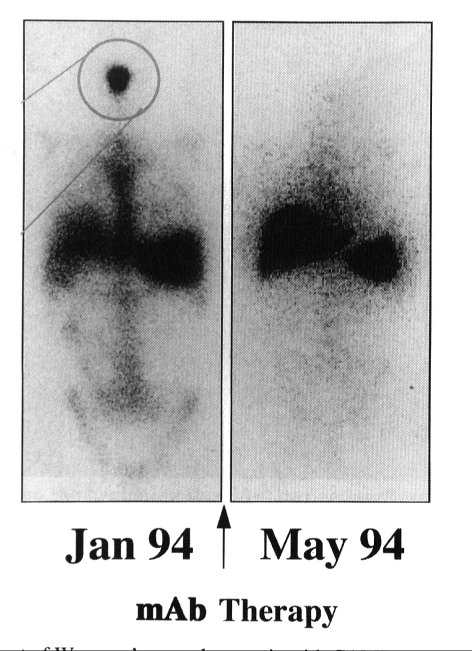
This patient suffered from severe erosion of sinue passages due to leukocyte infultration, as shown by the whole body scanning after injection of 111In-labelled leukocytes (area circled). Antibody treatment depleted the T cells which were driving the inflammatory process and this resulted in a normal leukocyte scan and disease stabilization for at least 12 months. (Reproduced with permission from Reuter et al. Quart. J. Med. 88, 509-516.)
Other autoimmune diseases were potential candidates for lymphocyte depletion therapy and in 1991, John Isaacs, a clinical research fellow in our group, started a collaboration with Brian Hazleman to treat patients with rheumatoid arthritis [29]. Severe disease carries a substantial morbidity and mortality in addition to the pain and progressive disablement, so many investigators believe that radical treatment is justifiable if could offer a long term benefit. The initial clinical responses to CAMPATH-1H were quite impressive and because of the obvious large market for a new treatment, Wellcome soon started a full-scale program of clinical trials in Europe and the USA. However, the benefit proved to be relatively short-lived (about three months) whereas a new and unpredicted problem emerged. Lymphocyte depletion by CAMPATH-1H was very profound and a single dose as low as 1 mg could reduce the blood lymphocyte count substantially. A typical course of treatment was 10 mg daily for 10 days and after this the T cell count remained near zero for several weeks. B cells and NK cells returned to normal levels by 8 weeks and CD8+ T cells followed more slowly. However, CD4+ T cells remained low, typically at about 200 per µl (about 20-40% of normal) [29,30]. There have been few studies on the functional significance of these low numbers, but it was reported that proliferative capacity to mitogens in vitro was reduced [30] and the T cell receptor repertoire might be diminished [31]. Nevertheless, immunity seems to be quite different from HIV-infected patients with similar low CD4+ numbers since there have been no cases of severe opportunistic infections or malignancies during the long-term follow up (apart from 4/172 in the first 6 months when T cells were most profoundly suppressed). These results were being documented by Wellcome in 1994 at about the same time as data was arriving from the trials in lymphoid malignancies. The majority of patients had been treated for advanced non-Hodgkin‘s lymphoma, mostly with bulky lymph nodes and resistant to multiple chemotherapy. As in our previous trials using CAMPATH-1G, the responses in lymph nodes were minimal. However, some small trials were done in chronic lymphocytic leukaemia, where the responses were more promising, and a particular subgroup of patients with T-cell proplymphocytic leukaemia (T-PLL) showed remarkable responses [32]. T-PLL is a rare but very aggressive disease which responds very poorly to chemotherapy and has a poor prognosis. At present, it is an unexplained paradox that this disease is so uniquely responsive to CAMPATH-1H.
In 1994 Wellcome reviewed their clinical data and decided that, notwithstanding the good results in leukaemia, there was likely to be insufficient commercial benefit for them to continue development of CAMPATH-1H. Of course this news was very disappointing to us, especially as the TAC was receiving requests every week from physicians around the world to supply CAMPATH-1 for transplant patients and patients with a range of severe auto-immune diseases. Large trials in bone marrow transplantation were showing significant benefits [33-35] and there were case reports of good results in treatment of cornea graft rejection [36], uveitis [37] scleroderma [38], and autoimmune cytopenias [39,40]. We were becoming very concerned how to support all these groups who clearly believed that CAMPATH-1 would help their patients.
One particularly interesting study was done by Alastair Compston‘s group at the Department of Neurology in Cambridge. In 1991 a middle-aged accountant with multiple sclerosis had insisted that something should be tried to arrest her progressive decline in mobility. By the time she received CAMPATH-1H, she was in a wheelchair. A few months later she went skiing! MS is a notoriously unpredictable disease and the placebo effect in clinical trials is considerable, so her improvement was only judged real when a series of gadolinium enhancing magnetic resonance imaging (MRI) scans showed that inflammation was significantly diminished. Her disease remained controlled for two years, providing sufficient encouragement to recruit a further six patients to a pilot study [41]. New lesions were again reduced and no serious adverse effects were seen, so a larger cohort was recruited. By 1998, 29 such patients have been treated with CAMPATH-1H and monitored for 18 months. The single treatment suppressed new MRI lesion formation by up to 90% compared to pre-treatment. Patients did not experience any attacks that might indicate new cerebral inflammation. More disappointing, though informative, is that about half of the patients have progressed. That is, the disability from pre-exisiting lesions has steadily got worse. It is concluded that the mechanism underlying this deterioration is non-inflammatory, possibly axonal degeneration following loss of the protective myelin sheath.
When this work started it was controversial whether MS was an autoimmune disease, but there was clearly significant immunopathology involving breakdown of the blood/brain barrier and lymphocyte infiltration. Whether antibody could reach these cells and change the course of the disease was unknown. Because of the great difficulty of measuring clinical effects, it was important to have a more objective disease marker. All of the patients had several MRI scans before treatment to establish the active and progressive nature of the lesions, followed by a series of scans afterwards. Again we should remark on the important contribution of these volunteers; although MRI scanning is non-invasive, it requires complete immobility in the claustrophobic core of the scanner for 20 minutes and is not a procedure anyone would enjoy. Many of them had to travel long distances for these scans on a regular basis.
Besides the reduction in new lesions detected by MRI, there were some completely unexpected results. We already knew that the first dose of CAMPATH-1H treatment was usually accompanied by a flu-like syndrome of fever, rigours, nausea, sometimes vomiting or marked hypotension. This is almost certainly caused by a rapid release of cytokines, including TNFa, IFNg and IL10, which has been well described for other anti-T cell antibodies like OKT3 [42]. It can be ameliorated by prior treatment with a pulse of corticosteroids. The reaction is self-limiting and is greatly diminished on the second and subsequent doses of antibody. The MS patients suffered the same syndrome, but it was accompanied by a dramatic, though fortunately short-lived, recapitulation or exacerbation of previous neurological symptoms [43]. At the same time, there was physiological evidence that nerve conduction had been blocked. Pre-treatment with steroids blocked both the "first-dose" syndrome and the neurologic effects. This suggested that one or more of the cytokines might have an impact on the previously damaged neurones and opened up new avenues for research into the pathology of MS.
The second unexpected complication was longer-term. As in other patients, CAMPATH-1H induced a long-lasting decrease in blood CD4+ T cell numbers. Between about 6 and 24 months post therapy about 30% of the patients developed anti-thyroid antibodies and hyperthyroidism (Graves‘ disease) [44]. There was no precedent for this in other patients who received CAMPATH-1H. Fortunately the clinical symptoms have been responsive to standard therapy. One hypothesis is that the antibody treatment may have brought about a shift in immune regulation eg from Th1 to Th2 type of response.
These clinical trials have helped to illuminate the underlying mechanisms of multiple sclerosis [45]. There seems to be a real possibility of arresting the inflammatory process. This gives an opportunity to test new therapies directed at re-myelination [46]. If this should involve tissue grafting, as is currently being considered, the immunosuppressive effect of CAMPATH-1H should still be helpful.
In parallel with the clinical research on CAMPATH-1 antibodies we spent many years working on the structure and possible function of the antigen. We wanted to know why it was such a good target for cell lysis so that similar therapeutic antibodies could be obtained against other cell types. The CAMPATH-1 antigen was very abundant, but antibodies against other highly expressed cell surface antigens like CD45 were by no means as lytic, particularly with human complement [47]. Conventional immunoprecipitation experiments with 125I or [ 35S]Met labelled protein were unsuccessful. However, we gave samples of antibody to several other scientists and it was a chance observation by Chris Barker which identified antigenic activity in chloroform/methanol extracts. This allowed antigen to be extracted and identified as a ladder of 20-25kD bands by SDS gel electrophoresis and Western blotting [48]. In 1987, Meng-Qi Xia joined our laboratory as a research student. She purified the antigen by affinity chromatography and gel electrophoresis and obtained 11 residues of N-terminal sequence. This was very short, but despite repeated efforts we could go no further. We had hoped to identify a peptide which would allow synthesis of an oligonucleotide probe for DNA cloning, but the sequences predicted were too short and redundant. A new postdoc in our lab, Masahide Tone, suggested that the redundant oligo might be used as a PCR primer along with a general poly-A primer to amplify the cDNA. Geoff was sceptical, but the experiment worked. We could hardly believe the DNA sequence, but it was confirmed by conventional cDNA cloning [49]. The surprising result was that the CAMPATH-1 antigen is a glycoprotein with only 12 amino acids. The C-terminal residue (Ser) was predicted to be attached to a glycosylphosphatidylinositol (GPI) anchor; that would account for why it was not detected by protein sequencing. We then had the good fortune to collaborate with Mike Ferguson, the doyen of GPI-anchorology and in his laboratory the whole structure of the anchor as well as the single complex N-linked oligosaccharide were determined [50,51].
As part of the pre-clinical testing of CAMPATH-1H, Wellcome had commissioned a study by Anthony Warford and Ian Lauder to determine its tissue cross-reactivity. They saw intense staining of mature sperm and specific epithelial cells in the male reproductive tract, as well as the expected reaction with lymphocytes and macrophages throughout the body [52]. It turned out that the CD52 antigen is produced by cells in the epididymis and seminal vesicle from where it is shed into the seminal fluid and acquired by sperm. The same result had been independently discovered by Christianne Kirchhoff, a reproductive biologist in Hamburg [53]. It is likely that the same antigen may have been examined by several teams who are looking for candidates for an intravaginal contraceptive [54]. We are often asked what is the function of the CD52 antigen. Its remarkable tissue distribution seems to be widely conserved. For some time we were spurred on by many discoveries of receptor-ligand pairs to look for a binding function. The best suggestion to date is from Neil Barclay, that considering its high abundance, extreme negative charge and cell distribution, the role of CD52 may be anti-adhesion, ie to keep the cells apart until the right time (since it is the unique role of sperm and lymphocytes to be able to migrate freely until they reach their target). As yet there are no data to support this proposition.
Following Wellcome‘s decision to abandon CAMPATH-1H, BTG approached a range of other companies to reach a new licence agreement, but without finding any interest. In the meantime, our group was in the process of moving to Oxford where we hoped to set up a new Therapeutic Antibody Centre. Its principal aim would still be to provide new biologicals for clinical research but now we were faced with the prospect of being the only source of CAMPATH-1 antibodies indefinitely, which was clearly beyond our resources or mandate. Wellcome (soon to be bought by Glaxo) were not longer able to support our work and so we urgently needed other industrial sponsorship. A link was made with LeukoSite Inc, a small new US Biotech company founded in 1993 by Tim Springer, who had worked with Herman at Cesar Milstein‘s laboratory in the late 1970s. Tim had pioneered the discovery of leukocyte adhesion molecules, including CD18, and this was the focus for the new company. Their chief executive, Chris Mirabelli took a bold risk when he committed a substantial proportion of the new company‘s start-up capital towards the construction and running of a new Centre for an academic group on the other side of the Atlantic. As our hope was fading that BTG would find a new licensee, LeukoSite became persuaded that CAMPATH-1H was a genuine opportunity, even though its first application (in chronic lymphocytic leukaemia), might be outside their original remit. There followed a long period of negotiation with BTG and Wellcome and a licence was finally agreed in April 1997. Thus began the next chapter in the story of CAMPATH-1.
Our experience has been a mixture of elation, despair and simple routine. We realised that it is never a simple process to develop a new pharmaceutical; many rounds of development and refinement may be needed to overcome each hurdle. This process was only made possible by a close relationship between the research group and the clinical teams. The ability of doctors in the UK to treat individual patients as they believe best and to conduct clinical trials under the DDX system (whereby they are exempt from the more onerous regulatory requirements) has been of inestimable value. Few, if any of the clinical developments described here could have been initiated by a pharmaceutical company. However, if we were not to abuse this system, it was important to have a production facility operating to the principles of Good Manufacturing Practice and following the relevant guidelines.
At the end of the day we know that only the pharmaceutical industry has the resources and expertise to bring a product to market. We need to work closely with them to transfer the technology and know-how in an effective way and to ensure that a fair (not extravagant) reward flows back to the academic institution when a potential product is marketed. In our experience it has been very much easier to interact with small biotech companies where the ethos is more akin to our academic culture and the management is closer to our level. To us, the big pharma like Glaxo/Wellcome seem daunting and impersonal; our main point of contact is with lawyers who appear obsessed with details we find trivial. We still don‘t know whether CAMPATH-1H will prove to be as widely useful for human therapy as we hope. We do know that it has been hugely enjoyable to be part of its development , sharing with literally thousands of scientists, physicians, nurses and patients worldwide. Along the way we have discovered many new aspects of therapeutic immunology relevant to other projects. We are just starting work in the lab on the next generation of CAMPATH-1 antibodies [55] and the physicians are continuing to find new applications [56].
We estimate that more than 2000 scientists, physicians, nurses and lawyers have been involved in the characterisation and development of CAMPATH-1 antibodies, and any success we have obtained is due to their cumulative collaborative efforts, as well as the incalculable contribution of over 3000 patients who have volunteered for clinical trials. However, we are solely responsible for the opinions expressed in this article. Over the last 20 years we have received financial support from the UK Medical Research Council, the Kay Kendall Leukaemia Fund, the Wellcome Foundation Ltd, LeukoSite Inc and several other charitable and industrial sources. GH is currently funded by the EP Abrahams‘ Trust.
1.von Behring, E., and Kitasato, S., (1890) Dtsch. Med. Wochenshe. 16, 1113-1114.
2.Hericourt, J. & Richet, Ch. (1895) "Physologie Pathologique" - de la serotherapie dans la traitement du cancer. Comptes Rendus Hebd. Seanc. Acad. Sci. 121, 567.
3.Currie, G.A., (1972) Eighty years of immunotherapy: a review of immunological methods used for the treatment of human cancer. Brit. J. Cancer 26, 141-153.
4.Prentice, H.G., Blacklock, H.A., Janossy, G., Bradstock, K.F., Skeggs, D., Goldstein, G. and Hoffbrand, A.V. (1982) Use of anti-T cell monoclonal antibody OKT3 to prevent acute graft versus host disease in allogeneic bone marrow transplantation for acute leukemia. Lancet 1, 700-703.
5.Filipovitch, A.H., Vallera, D.A., Youle, R.J., Haake, R., Blazar, B.R., Arthur, D., Neville, Ramsay, N.K., McGlave, P. and Kersey, J.H. (1987) Graft-versus-host disease prevention in allogeneic bone marrow transplantation from hitocompatible siblings. A pilot study using immunotoxins for T cell depletion of donor bone marrow. Transplantation 44, 62-69.
6.Clark, M., Cobbold, S., Hale, G., and Waldmann, H. (1983) Advantages of rat monoclonal antibodies. Immunol. Today 4, 100-101.
7.Hale, G., Bright, S., Chumbley, G., Hoang, T., Metcalf, D., Munro, A.J., Waldmann, H. (1983) Removal of T cells from bone marrow for transplantation: a monoclonal antilymphocyte antibody that fixes human complement. Blood 62, 873-882.
8.Hale, G., Hoang, T., Prospero, T., Watt, S.M., and Waldmann, H. (1983) Removal of T cells from bone marrow for transplantation: comparison of rat monoclonal anti-lymphocyte antibodies of different isotypes. Mol. Biol. Med. 1, 305-319.
9.Hale, G., Swirsky, D.M., Hayhoe, F.G.J., and Waldmann, H. (1983) Effects of monoclonal anti-lymphocyte antibodies in vivo in monkeys and humans. Mol. Biol. Med. 1, 321-334.
10.Waldmann, H., Or, R., Hale, G., Weiss, L., Cividalli, G., Samuel, S., Manor, D., Brautbar, C., Polliack, A., Rachmilewitz, E.A., and Slavin, S. (1984) Elimination of graft versus host disease by in vitro depletion of alloreactive lymphocytes using a monoclonal rat anti-human lymphocyte antibody (CAMPATH-1). Lancet 2, 483-486.
11.Heit, W., Bunjes, D., Weisneth, M., Schmeiser, T., Arnold, R., Hale, G., Waldmann, H., and Heimpel, H. (1986) Ex vivo T-cell depletion with the monoclonal antibody Campath-1 plus human complement effectively prevents acute GvHD in allogeneic bone marrow transplantation. Brit. J. Haematol. 64, 479-486.
12.Goldman, J.M., Apperley, J.F., Jones, L., Marcus, R., Goolden, A.W.G., Batchelor, R., Hale, G., Waldmann, H., Reid, C.D., Hows, J., Gordon-Smith, E., Catovsky, D., and Galton, D.A.G. (1986) Boe marrow transplantation for patients with chronic myeloid leukemia. New Engl. J. Med. 314, 202-207.
13.Hale, G., Cobbold, S., Waldmann, H. (1988) T cell depletion with CAMPATH-1 in allogeneic bone marrow transplantation. Transplantation 45, 753-759.
14.Cobbold, S.P., Thierfelder, S. and Waldmann, H. (1983). Immunosuppression with Monoclonal Antibodies: A Model to Determine the Rules for Effective Serotherapy. Mol. Biol. Med. 1, 285-304.
15.Cobbold, S.P., Martin, G. and Waldmann, H. (1986). Monoclonal antibodies for the prevention of graft-versus-host disease and marrow graft rejection: The depletion of T-cell subsets in vitro and in vivo1. Transplantation 42, 239-247.
16.Hale, G., Clark, M., and Waldmann, H. (1985) Therapeutic potential of rat monoclonal antibodies: Isotype specificity of antibody-dependant cell-mediated cytotoxicity with human lymphocytes. J. Immunol. 134, 3056-3061.
17.Bruggemann, M., Free, J., Diamond, A., Howard, J., Cobbold, S.P. and Waldmann, H. (1986). Immunoglobulin heavy chain locus of the rat: Striking homology to mouse antibody genes. Proc. Natl. Acad. Sci. 83, 6075-6079.
18.Hale, G., Cobbold, S.P., Waldmann, H., Easter, G., Matejtschuk, P., and Coombs, R.R.A. (1987) Isolation of low-frequency class-switch variants from rat hybrid myelomas. J. Immunol. Meth. 103, 59-67.
19.Dyer, M.J.S., Hale, G., Hayhoe, F.G.J., and Waldmann, H. (1989) Effects of CAMPATH-1 antibodies in vivo in patients with lymphoid malignancies: influence of antibody isotype. Blood 73, 1431-1439.
20.Neuberger, M.S., Williams, G.T., Mitchell, E.B., Jouhal, S.S., Flanagan, J.G., and Rabbitts, T.N. (1985) a hapten-specific chimaeric IgE antibody with human physiological effector function. Nature 314, 268-270.
21.Jones, P.T., Dear, P.H., Foote, J., Neuberger, M.S., and Winter, G. (1986) Replacing the complementarity-determining regions in a human antibody with those from a mouse. Nature 321, 522-525.
22.Friend, P.J., Waldmann, H., Hale, G., Cobbold, S., Rebello, P., Thiru, S., Jamieson, N.V., Johnston, P.S., & Calne, R.Y. (1991) Reversal of allograft rejection using the monoclonal antibody CAMPATH-1G. Transplant. Proc. 23, 1390-1392.
23.Riechmann, L. Clark, M., Waldmann, H., and Winter, G. (1988). Reshaping human antibodies for therapy. Nature 332, 323-327.
24Bruggemann, M., Williams, G.T., Bindon C.I., Clark, M.R., Walker, M.R., Jefferies, R., Waldmann, H. & Neuberger, M.S. (1987). Comparison of the Effector Functions of Human Immunoglobulins Using a Matched Set of Chimeric Antibodies. J. Exp. Med. 166, 1351-1361.
25.Hale, G., Dyer, M.J.S., Clark, M.R., Phillips, J.M., Marcus, R., Riechmann, L., Winter, G., and Waldmann, H. (1988) Remission induction in non-Hodgkin lymphoma with reshaped human monoclonal antibody CAMPATH-1H. Lancet 2, 1394-1399.
26.Hale, G. (1993) Small scale production of novel therapeutic products: A new challenge for the transfusion service? (editorial) Transfus. Med. 3, 1-5.
27.Lockwood, C.M., Thiru, S., Isaacs, J.D., Hale, G., and Waldmann, H. (1993) Humanised monoclonal antibody treatment for intractable systemic vasculitis. Lancet 341, 1620-1622.
28.Lockwood, C.M., Thiru, S., Stewart, S., Hale, G., Isaacs, J.D., Wraight, P., Elliott, J. and Waldmann, H. (1996) Treatment of refractory Wegener‘s granulomatosis with humanised monoclonal antibodies. Q. J. Med 89, 903-912.
29.Isaacs, J.D., Watts, R.A., Hazleman, B.L., Hale, G., , Keogan, M.T., Cobbold, S.P., and Waldmann, H. (1992) Humanised monclonal antibody therapy for rheumatoid arthritis. Lancet, 340, 748-752.
30.Brett, S., Baxter, G., Cooper, H., Johnston, J.M., Tite, J., and Rapson, N. (1996) Repopulation of blood lymphocyte sub-populations in rheumatoid arthritis patients treated with the depleting humanized monoclonal antibody, CAMPATH-1H. Immunology 88, 13-19.
31.Jendro, M.C., Ganten, T., Matteson, E.L., Weyand, C.M., and Goronzy, J.J., (1995) Emergence of ologoclonal T cell populations following therapeutic T cell depletion in rheuamtoid arthritis. Arth. Rheum. 38, 1242-1251.
32.Pawson, R., Dyer, M.J.S., Barge, R., Matutes, E., Thornton, P.D., Emmett, E., Kluin-Nelemans, J.C., Fibbe, W.E., Willemze, R., and Catovsky, D. (1997) Treatment of T-cell prolymphocytic leukaemia with human CD52 antibody. J. Clin. Oncol. 15, 2667-2672.
33.Jacobs, P. Wood, L., Fullard, L., Waldmann, H. and Hale, G. (1993) T-cell depletion by exposure to CAMPATH-1G in vitro prevents graft-versus-host disease. Bone Marrow Transplantation. 13, 763-769.
34.Hale, G., and Waldmann, H. for CAMPATH users (1994) Control of graft-versus-host disease and graft rejection by T cell depletion of donor and recipient with CAMPATH1 antibodies. Results of matched sibling transplants for malignant diseases. Bone Marrow Transplantation 13, 597-611.
35.Hale, G., and Waldmann, H. for CAMPATH users (1994) CAMPATH1 monoclonal antibodies in bone marrow transplantation. Hematotherapy 3, 15-31.
36.Newman, D.K., Isaacs, J.D., Watson, P.G., Meyer, P.A., Hale, G., and Waldmann, H. (1995) Prevention of immune-mediated corneal graft destruction with the anti-lymphocyte monoclonal antibody, CAMPATH-1H. Eye 9, 564-569.
37.Isaacs, J.D., Dick, A.D., Haynes, R., Watson, P., Forrester, J.V., Myer, P., Hale, G., and Waldmann, H. (1996) Monoclonal antibody therapy of chronic intraocular inflammation using CAMPATH-1H. Brit. J. Opthalmol. 79, 1054-1055.
38.Isaacs, J.D., Hazleman, B.L., Chakravarty, K., Grant, J.W., Hale, G., and Waldmann, H. (1996) Monoclonal antibody therapy of diffuse cutaneous scleroderma with CAMPATH-1H. J. Rheumatol. 23, 1103-1106.
39.Lim, S.H., Hale, G., Marcus, R.E., Waldmann, H and Baglin, T.P. (1993) CAMPATH-1 MoAb in the treatment of refractory autoimmune thrombocytopenic purpura Brit. J. Haematol. 84, 542-544.
40.Killick, S.B., Marsh, J.C.W., Hale, G., Waldmann, H., Kelly, S.J., and Gordon-Smith, E.C. (1997) Sustained remission of severe resistant auto-immune neutropenia with Campath1H. Brit J. Haematol. 97, 306-308.
41.Moreau, T., Thorpe, J., Miller, D., Moseley, I., Hale, G., Waldmann, H., Clayton, D., Wing, M., Scolding, N. and Compston, A. (1994) Preliminary evidence from magnetic resonance imaging for reduction in disease activity after lymphocyte depletion in multiple sclerosis. Lancet 344, 298-301.
42.Chatenoud L, Ferran C, Legendre C, Thouard I, Merite S, Reuter A, Gevaert Y, Kreis H, Franchimont P & Bach J-F (1990) In vivo cell activation following OKT3 adminstration. Systematic cytokine release and modulation by corticosteroids. Transplantation 49, 697-702.
43.Moreau, T., Coles, A., Wing, M.G., Isaacs, J., Hale, G., Waldmann, H., and Compston, A., (1996) Transient increase in symptoms associated with cytokine release in patients with multiple sclerosis Brain 119, 225-237.
44.Coles, A.J., Wing, M.G., Corradu, F., Smith, S.I., Taylor, C.J., Hale, G., Waldmann, H., Weetman, A.P., Chatterjee, V.K., & Compston, A. (1998) Pulsed monoclonal antibodies treatment modulates T cell responses in multiple sclerosis but induces autoimmune thyroid disease. manuscript in preparation.
45.Coles, A.J., Paolili, A., Molyneux, P., Wing, M.G., Hale, G., Miller, D. Waldmann, H. & Compston, A. (1998) Monoclonal antibody treatment exposes three mechanisms underlying the clinical course of multiple sclerosis. manuscript in preparation.
46.Compston, A. (1994) Future prospects for the management of multiple sclerosis. Ann Neurol. 36, S146-S150.
47.Bindon, C.I., Hale, G., and Waldmann, H. (1988) Importance of antigen specificity for complement mediated lysis by monoclonal antibodies. Eur. J. Immunol. 18, 1507-1514.
48.Hale, G., Xia, M-Q., Tighe, H.P., Dyer, M.J.S. and Waldmann, H. (1990) The CAMPATH-1 antigen (CDw52). Tissue Antigens 35, 118-127.
49.Xia, M-Q., Tone, M., Packman, L., Hale, G. and Waldmann, H. (1991) Characterization of the CAMPATH-1 antigen: biochemical analysis and cDNA cloning reveal an unusually small peptide backbone. Eur. J. Immunol. 21, 1677-1684.
50.Xia, M-Q., Hale, G., Lifely, M.R., Ferguson, M.J., Campbell, D., Packman, L. and Waldmann, H. (1993) Structure of the CAMPATH-1 antigen, a GPI-anchored glycoprotein which is an exceptionally good target for complement lysis. Biochem. J. 293, 633-640.
51.Treumann, A., Lifely, R., Schneider, P., and Ferguson, M.A.J. (1995) Primary structure of CD52. J. Biol. Chem. 270, 60886099.
52.Hale, G., Rye, P.D., Warford, A., Lauder, I., and Brito-Babapulle, A. (1993) The GPI-anchored lymphocyte antigen CDw52 is associated with the epididymal maturation of human spermatozoa. J. Reprod. Immunol. 23, 189-205.
53.Kirchhoff, C. Krull, N., Pera, I., and Ivell, R. (1992) A major mRNA of the human epididymal principal cells, HE5, encodes the leucocyte differentiation CDw52 antigen peptide backbone. Mol. Repr. Dev. 34; 1115.
54.Diekman, A.B., Westbrook-Case, A., Naaby-Hansen, S., Klotz, K.L., Flickinger, C.J., and Herr, J.C. (1997) Biochemical characterization of sperm agglutination antigen-1, a human sperm surface antigen implicated in gamete interactions. Biol. Reprod. 57; 1136-1114.
55.Gilliland, L.K., Walsh, LA., Frewin, M.R., Wise, M., Tone, M., Hale, G., Kioussis, D. & Waldmann, H. (1998) Elimination of the immunogenicity of therapeutic antibodies. J. Immunol. 162: 3663-3671
56.Calne, R., Friend, P., Moffatt, S., Waldmann, H., Hale, G., Alexander, G., Jamieson, N & Firth, J. (1998) "Prope" tolerance: peri-operative CAMPATH-1H and low dose cyclosporin monotherapy in liver and renal allograft recipients. Lancet published.
This version converted to HTML by Steve Cobbold, 3rd April 2001