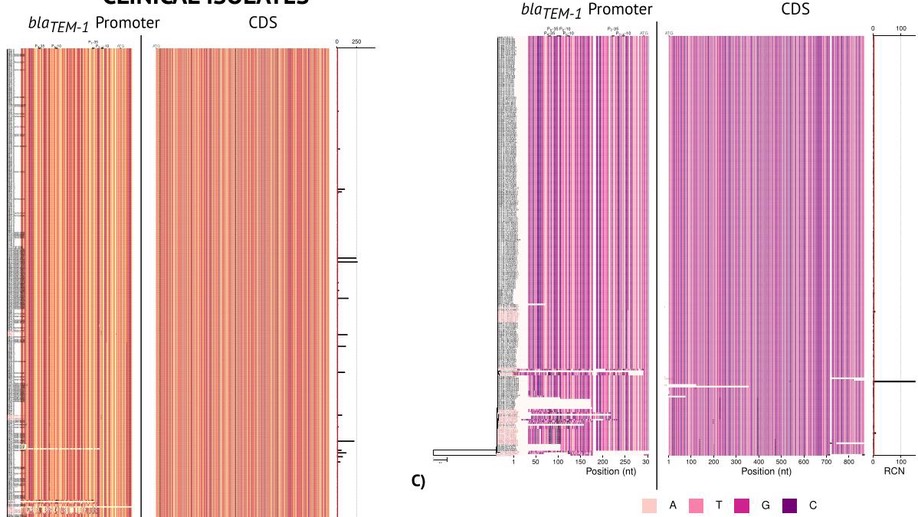
Hound: A novel tool for automated mapping of genotype to phenotype in bacterial genomes assembled de novo
Increasing evidence suggests that microbial species have a strong within species genetic heterogeneity. This can be problematic for the analysis of prokaryote genomes, which commonly relies on a reference genome to guide the assembly process: any difference between reference and sample genomes can introduce errors in the detection of small insertions, deletions, structural variations and even point mutations. This phenomenon jeopardises the genomic surveillance of antibiotic-resistant bacteria, triggering even a reproducibility crisis. Here we present Hound, an analysis pipeline that integrates publicly available tools to locally assemble prokaryote genomes de novo, detect genes by similarity, and report the mutations found. Three features are exclusive to Hound: It reports relative gene copy number, retrieves sequences upstream the start codon to detect mutations in promoter regions, and, importantly, can merge contigs based on a user-given query sequence to reconstruct genes that are fragmented by the assembler. To demonstrate Hound, we screened through 5,032 bacterial whole-genome sequences isolated from farm animals and clinical patients using the amino acid sequence of blaTEM-1, to predict resistance to amoxicillin-clavulanate. We believe this tool can facilitate the analysis of prokaryote species that currently lack a reference genome, and can be scaled up to build automated diagnostic systems.
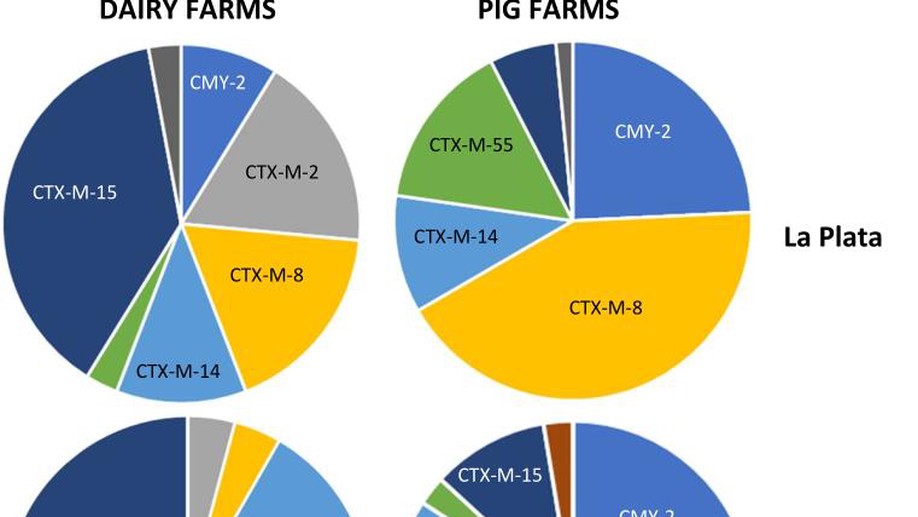
Genomic epidemiology of third-generation cephalosporin-resistant Escherichia coli from Argentinian pig and dairy farms reveals animal-specific patterns of co-resistance and resistance mechanisms
Little is known about the ecology of critically important antibiotic resistance among opportunistic human pathogens (e.g. Escherichia coli) on South American farms. By studying 70 farms in central-eastern Argentina, we identified that third-generation cephalosporin resistance (3GC-R) in E. coli was mediated by mechanisms seen more often in certain species (pigs or dairy cattle) and that 3GC-R pig E. coli were more likely to be co-resistant to florfenicol and amoxicillin/clavulanate. This suggests that on-farm antibiotic usage is key to selecting the types of E. coli present on these farms. 3GC-R E. coli were highly phylogenetically variable and we identified the de novo mobilisation of the resistance gene blaROB, alongside a novel florfenicol resistance gene, from pig pathogens into E. coli on a mobile genetic element that was widespread in the study region. Overall, this shows the importance of surveying poorly studied regions for critically important antibiotic resistance which might impact human health.

Improving Nitrofurantoin Resistance Prediction in Escherichia coli from Whole Genome Sequence by Integrating NfsA/B Enzyme Assays
Nitrofurantoin resistance in Escherichia coli is primarily caused by mutations damaging two enzymes, NfsA and NfsB. Studies based on small isolate collections with defined nitrofurantoin MICs have found significant random genetic drift in nfsA and nfsB making it extremely difficult to predict nitrofurantoin resistance from whole genome sequence (WGS) where both genes are not obviously disrupted by nonsense or frameshift mutations or insertional inactivation. Here we report a WGS survey of 200 E. coli from community urine samples, of which 34 were nitrofurantoin resistant. We characterised individual non-synonymous mutations seen in nfsA and nfsB among this collection using complementation cloning and assays of NfsA/B enzyme activity in cell extracts. We definitively identified R203C, H11Y, W212R, A112E, A112T and A122T in NfsA and R121C, Q142H, F84S, P163H, W46R, K57E and V191G in NfsB as amino acid substitutions that reduce enzyme activity sufficiently to cause resistance. In contrast, E58D, I117T, K141E, L157F, A172S, G187D and A188V in NfsA and G66D, M75I, V93A and A174E in NfsB, are functionally silent in this context. We identified that 9/166 (5.4%) of nitrofurantoin susceptible isolates were pre-resistant, defined as having loss of function mutations in nfsA or nfsB. Finally, using NfsA/B enzyme activity assay and proteomics we demonstrated that 9/34 (26.5%) of nitrofurantoin resistant isolates carried functionally wild-type nfsB or nfsB/nfsA. In these cases, enzyme activity was reduced through downregulated gene expression. Our biological understanding of nitrofurantoin resistance is greatly improved by this analysis, but is still insufficient to allow its reliable prediction from WGS data.
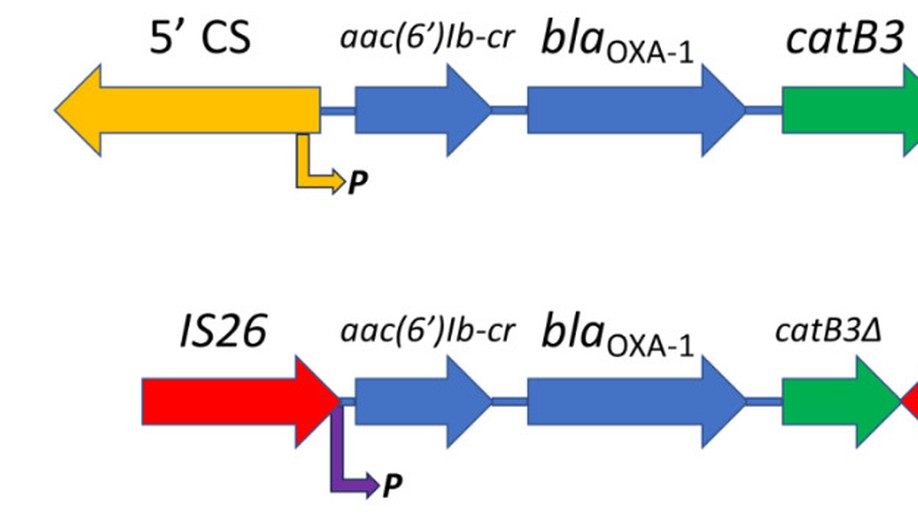
Harvesting and amplifying gene cassettes confers cross-resistance to critically important antibiotics
Amikacin and piperacillin/tazobactam are frequent antibiotic choices to treat bloodstream infection, which is commonly fatal and most often caused by bacteria from the family Enterobacterales. Here we show that two gene cassettes located side-by-side in and ancestral integron similar to In37 have been “harvested” by insertion sequence IS26 as a transposon that is already globally disseminated among the Enterobacterales. This transposon encodes the enzymes AAC(6’)-Ib-cr and OXA-1, reported, respectively, as amikacin and piperacillin/tazobactam resistance mechanisms. However, by studying bloodstream infection isolates from 769 patients from three hospitals serving a population of 1.5 million people in South West England, we show that increased enzyme production due to mutation in an IS26/In37-derived hybrid promoter or, more commonly, transposon copy number amplification is required to simultaneously remove these two key therapeutic options; in many cases leaving only the last-resort antibiotic, meropenem. These findings may help improve the accuracy of predicting piperacillin/tazobactam treatment failure, allowing stratification of patients to receive meropenem or piperacillin/tazobactam, which may improve outcome and slow the emergence of meropenem resistance.

Predicting the re‐distribution of antibiotic molecules caused by inter‐species interactions in microbial communities
Drug efficacy measured using pure cultures is not maintained in the presence of neighbouring microbes. Using simple physical laws, I pinpoint the mechanism for this change to demonstrate that drug efficacy can be predictably manupulated, and suggest why microbes began to co-operate and form communities.

Plasmid carriage and the unorthodox use of Fisher's theorem in evolutionary biology
The link between fitness and reproduction rate is a central tenet in evolutionary biology: mutants reproducing faster than the dominant wild-type are favoured by selection, otherwise the mutation is lost. This link is given by Fisher’s theorem under the assumption that fitness can only change through mutations. Here I show that fitness, as formalised by Fisher, changes through time without invoking new mutations—allowing the co-maintenance of fast- and slow-growing genotypes. The theorem does not account for changes in population growth that naturally occur due to resource depletion, but it is key to this unforeseen co-maintenance of growth strategies. To demonstrate that fitness is not constant, as Fisher’s theorem predicates, I co-maintained a construct of Escherichia coli harbouring pGW155B, a non-transmissible plasmid that protects against tetracycline, in competition assays without using antibiotics. Despite growing 40% slower than its drug-sensitive counterpart, the construct with pGW155B persisted throughout the competition without tetracyclin—maintaining the plasmid. Thus, predicting the selection of microbial genotypes may be more difficult than previously thought.
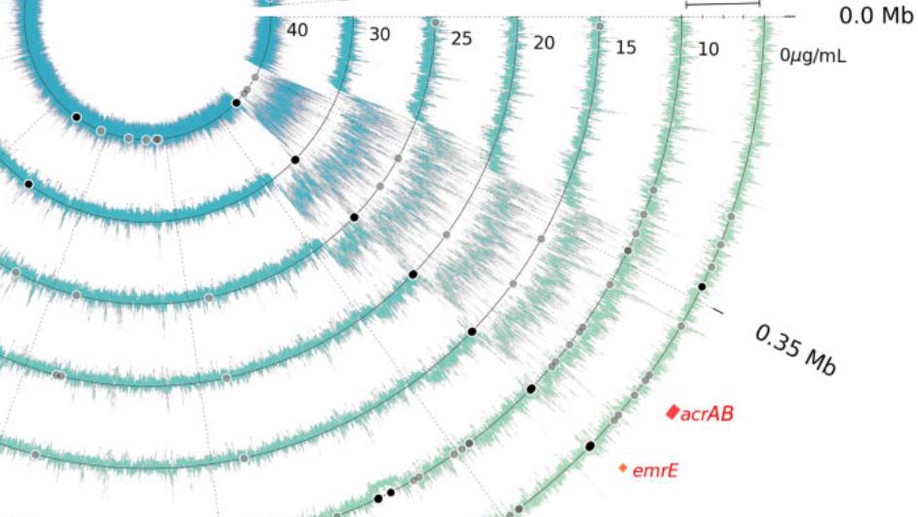
The Antibiotic Dosage of Fastest Resistance Evolution: Gene Amplifications Underpinning the Inverted-U
The Mutant Selection Window (MSW) claims that selection for resistance begins only after the minimum inhibitory concentration, but there is no rationale behind it and no data to support it. Here we exposed E. coli to different antibiotic concentrations to measure which leads to fastest adaptation, and underpin the genetic mechanism.

Image-based spectrophotometer: The Light-Modulator (LiMO®)
We dreamt of phenotyping microbes and running evolution experiments with antibiotics at much higher-throughput, using many more strains and conditions than before.
The solution had to be cheap, small, portable and accurate. So, with ERC funding, we set about leveraging low-cost robotics, adapting some LED lighting technology and using our own algorithm-building expertise to create the kind of device we wished we could buy.

Canonical host-pathogen tradeoffs subverted by mutations with dual benefits
Tradeoffs between life history traits impact diverse biological phenomena, including the maintenance of biodiversity. We sought to study two canonical tradeoffs in a model host-parasite system consisting of bacteriophage lambda and Escherichia coli: i) parasite resistance for growth and ii) phage infectivity for host-range. We report that these previously hypothesised tradeoffs are, in fact, tradeups. While the observation of tradeups was surprising, they should be expected because if traits X and Y tradeoff, so too traits Y and Z, then X and Z will tradeup. By considering five different E. coli trait correlations we uncovered several tradeups and tradeoffs. Using mathematical models, we establish that tradeups need not inhibit biodiversity, as previously thought, and can help maintain it through high-dimensional trait interactions. We provide a mechanistic explanation for how tradeups emerge and give reasons for why tradeups can even evolve in well-adapted genomes.

Fluorescence photography of patterns and waves of bacterial adaptation at high antibiotic doses
Fisher suggested advantageous genes would spread through populations as a wave so we sought genetic waves in evolving populations, as follows. By fusing a fluorescent marker to a drug efflux protein (AcrB) whose expression provides Escherichia coli with resistance to some antibiotics, we quantified the evolution and spread of drug-resistant E. coli through spacetime using image analysis and quantitative PCR. As is done in hospitals routinely, we exposed the bacterium to a gradient of antibiotic in a ‘disk diffusion’ drug susceptibility test that we videoed. The videos show complex spatio-genomic patterns redolent of, yet more complex than, Fisher’s predictions whereby a decelerating wave front of advantageous genes colonises towards the antibiotic source, forming bullseye patterns en route and leaving a wave back of bacterial sub-populations expressing acrB at decreasing levels away from the drug source. qPCR data show that E. coli sited at rapidly-adapting spatial hotspots gain 2 additional copies of acr, the operon that encodes AcrB, within 24h and imaging data show resistant sub-populations thrive most near the antibiotic source due to non-monotone relationships between inhibition due to antibiotic and distance from the source. In the spirit of Fisher, we provide an explicitly spatial nonlinear diffusion equation that exhibits these properties too. Finally, linear diffusion theory quantifies how the spatial extent of bacterial killing scales with increases in antibiotic dosage, predicting that microbes can survive chemotherapies that have been escalated to 250× the clinical dosage if the antibiotic is diffusion-limited.

The unconstrained evolution of fast and efficient antibiotic-resistant bacterial genomes
For decades it was hypothesised that the growth rate of populations and their size engage in a trade-off, but the data was always inconclusive. Using bacteria, we set off to underpin a physical mechanism for this trade-off, and then explain why it is not always found in the data.

Using a Sequential Regimen to Eliminate Bacteria at Sublethal Antibiotic Dosages
Lorem ipsum dolor sit amet, consectetur adipiscing elit. Duis posuere tellus ac convallis placerat. Proin tincidunt magna sed ex sollicitudin condimentum.
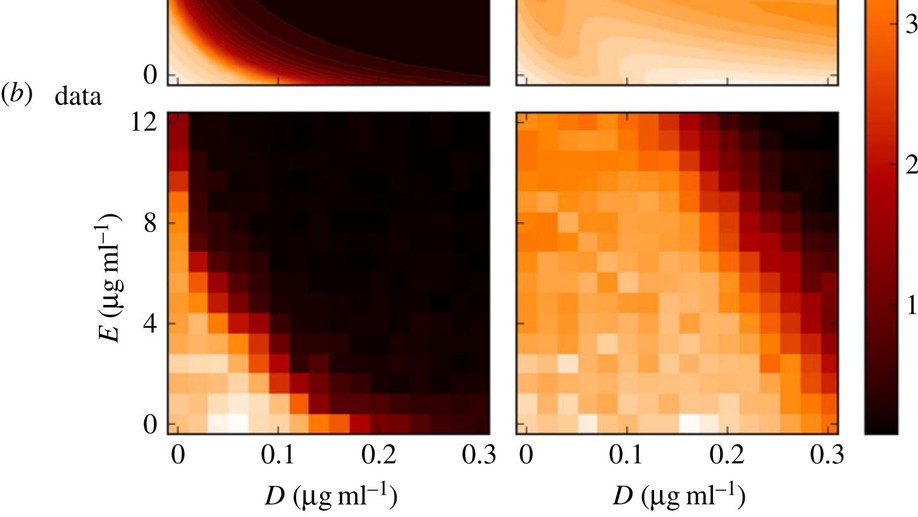
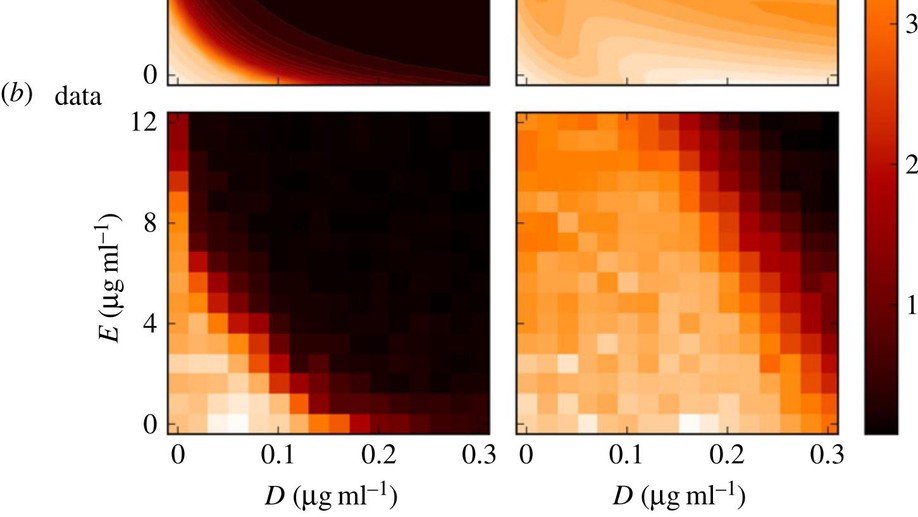
Testing the optimality properties of a dual antibiotic treatment in a two-locus, two-allele model
Mathematically speaking, it is self-evident that the optimal control of complex, dynamical systems with many interacting components cannot be achieved with ‘non-responsive’ control strategies that are constant through time. Although there are notable exceptions, this is usually how we design treatments with antimicrobial drugs when we give the same dose and the same antibiotic combination each day. Here, we use a frequency- and density-dependent pharmacogenetics mathematical model based on a standard, two-locus, two-allele representation of how bacteria resist antibiotics to probe the question of whether optimal antibiotic treatments might, in fact, be constant through time. The model describes the ecological and evolutionary dynamics of different sub-populations of the bacterium Escherichia coli that compete for a single limiting resource in a two-drug environment. We use in vitro evolutionary experiments to calibrate and test the model and show that antibiotic environments can support dynamically changing and heterogeneous population structures. We then demonstrate, theoretically and empirically, that the best treatment strategies should adapt through time and constant strategies are not optimal.
Regeneración retiniana asociada a hidrocefalia congénita debida a mutación en el gen que codifica alfa-SNAP
En el ratón mutante hyh existen bajos niveles funcionales de la proteína alfa-SNAP para interaccionar con el complejo SNARE implicado en el tráfico vesicular intracelular, afectándose el proceso de secreción y adhesión celular durante al desarrollo del sistema nervioso central. Consecuentemente, los ratones mutantes manifiestan hidrocefalia congénita por desprendimiento del neuroepitelio, probablemente debido a alteraciones en la adhesión. Algunos síndromes humanos presentan hidrocefalia asociada a defectos retinianos, como el síndrome de Walker-Waburg, con displasia retiniana. La hidrocefalia del ratón hyh ha demostrado ser un modelo representativo de la humana, y por eso el estudio de su degeneración retiniana asociada puede arrojar luz sobre defectos parecidos en la hidrocefalia humana. Se han estudiado las retinas de ratones mutantes con la enfermedad cronificada que han podido sobrevivir hasta edades avanzadas. Las retinas de ratones normales y mutantes, desde el nacimiento hasta 180 días postnatales, se procesaron para reconocer sus distintos tipos celulares e identificar los que mueren. Se estudió la expresión de alfa-SNAP y la implicación del citoesqueleto y moléculas de adhesión en la degeneración, así como el papel que desempeña en el proceso degenerativo la microglía/macrófagos y los astrocitos. Se encontraron diversos defectos en las retinas de los animales hidrocefálicos, consistentes en muertes de fotorreceptores y otros tipos celulares, destacando la ausencia casi total de conos y bastones y de la capa plexiforme externa.