This page presents a summary of the work I have been involved with since I first entered the field of diabetes research.
The substrate-site hypothesis
The underlying theme of my research had its origins in 1964 when Hal Coore and Philip Randle used the newly invented radioimmunoassay to carry out the first in vitro studies on insulin secretion. From the specificity of the secretory responses to sugars they were led to suggest that “...the phosphorylation of glucose...within the ß-cell may provide the signal for insulin release induced by glucose”. This we later formulated more precisely in terms of two possible models for glucose-induced insulin release. In the first of these which we termed the Regulator site hypothesis glucose was envisaged as binding to a specific glucoreceptor akin to a hormone or neurotransmitter receptor to initiate changes leading to secretion of insulin. In the second, termed the Substrate site hypothesis, the trigger for secretion is produced from the metabolism of the sugar within the ß-cell.
The first phase of studies on the substrate-site hypothesis were aimed at establishing the validity of the substrate-site hypothesis. The substantive observations were that rates of glucose metabolism showed the same sigmoidal dependence on glucose concentration to that shown by glucose-stimulated insulin release; the specificity of the two processes is the same, only metabolisable sugars stimulating insulin release; inhibition of glucose metabolism produces a corresponding inhibition of insulin release; and other fuel molecules that can enter ß-cell metabolism also stimulate insulin release.
It followed that the enzymic step(s) responsible for controlling glucose metabolism could be equated with the ‘glucoreceptor’. In many mammalian tissues transport of the sugar is rate-limiting for its metabolism. The concentration of the sugar within the cell is therefore much lower than in the blood and phosphorylation is carried out by a low Km hexokinase. Such is not the case for the ß-cell in which the situation is comparable to that in liver - i.e. rapid non-rate-limiting transport of the sugar so that there is equilibration across the plasma membrane followed by rate-limiting phosphorylation to glucose 6-phosphate catalysed by a high Km glucokinase. There is also evidence for the presence of G6Pase catalysing the reverse reaction to make the step potentially a substrate cycle - the quantitative importance of the G6Pase is however open to question. The central importance of glucokinase, however, is unquestionable. Its high Km and specificity for glucose account for the key properties of glucose-stimulated insulin release; glucokinase unusually shows sigmoid kinetics despite being monomeric and this is important for the threshold response to blood glucose concentrations just above starving levels; application of Metabolic Control Analysis to ß-cell glycolysis confirms the high control strength of glucokinase; its inhibition by mannoheptulose has been an important tool for exploring the scope of the Substrate Site hypothesis; and mutations in glucokinase can result in disorder of insulin secretion in MODY and HI.
It is always the apparent exceptions that provide the challenge for a theory. Three exceptions proved the rule for the Substrate Site hypothesis. Firstly N-acetylglucosamine (NAG) was found to stimulate insulin release from rat islets but is NOT a substrate for GK - this was resolved when it was found that rat islets contain a specific NAG kinase which allows NAG to enter glycolysis. Secondly, non-metabolisable leucine analogues e.g. BCH were found to have effects on insulin secretion similar to those of leucine itself - this is resolved by the demonstration that a non-metabolisable AA can markedly modify endogenous metabolism e.g. by activating GDH and thereby elicit metabolism-dependent effects. Finally, in HIT T15 cells we discovered that mannoheptulose did not inhibit glucose-stimulated insulin release. However this problem for the Substrate Site hypothesis had its resolution in the demonstration that in HIT T15 cells the rate-limiting step for glucose uptake is not glucokinase but rather is the preceding step of glucose transport - MH does not compete effectively with glucose for uptake and this does not enter the cell to inhibit GK.
Three classes of signals affected the ß-cell can be distinguished: initiators which are capable of stimulating insulin release in the absence of other agents; potentiators which amplify the effect of an initiator; and inhibitors. Potentiators and many inhibitors are hormones and neurotransmitters which activate cell surface receptors and modulate classical second messenger pathways. Initiators fall into two groups - metabolites such as glucose, and non-metabolites in particular the sulfonylureas. The unifying concept is that both initiator groups ultimately affect insulin secretion by leading to an increase in ß-cell [Ca]. Both groups increase [Ca] by causing ß-cell membrane depolarisation which allows Ca-entry through voltage-sensitive Ca-channels. In addition, glucose and other metabolites mobilise intracellular Ca from intracellular stores. The former mechanism is now understood in molecular detail . The link between metabolism and Ca-mobilisation, however, remains much less well defined.
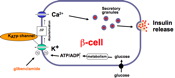
How does glucose cause membrane depolarisation? The first clear indication of the mechanism linking glucose metabolism to cellular membrane potential came from an experiment carried out by myself, Donna Harrison and Frances Ashcroft in which we showed for the first time that a specific ß-cell K-channel could be blocked by glucose and that this effect could be reversed by mannoheptulose hence implicating glucose metabolism in K-channel closure.
The Substrate Site hypothesis makes no prediction as to the link between metabolism of glucose and the cell depolarisation which leads to Ca-influx and insulin release. However it was soon shown that the K-channel that is closed in response to an increase in metabolic flux is an ATP-sensitive K-channel, (KATP channel) which closes in response to an increase in ATP, and opens in response to an increase in ADP. The KATP channel is also the site of action of sulfonylureas used in the treatment of non-sinsulin dependent diabetes; drugs of this class bind to the channel and cause it to close. Thus the link between glucose metabolism and triggering of insulin secretion by an influx of Ca ions is as follows - an increase in blood glucose leading to increased [glucose] within the ß-cell and increased phosphorylation and subsequent metabolism of the sugar. The increased metabolic flux causes an increase in [ATP]/[ADP], closure of the KATP channel and consequent depolarisation of the cell membrane. The ensuing opening of voltage-sensitive Ca-channels leads to Ca-influx, an increase in [Ca]
What do we know about the effects of nucleotides on the channel? A major advance was the cloning of a channel subunit and the demonstration that the channel contains two subunits - Kir6.2, the channel protein itself, and SUR1 which acts as a channel regulator and mediates sensitivity to sulfonylureas.
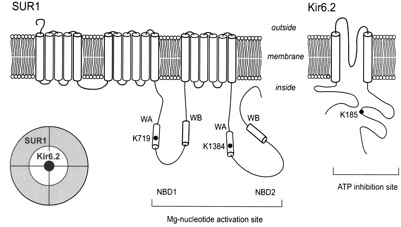
Kir6.2 has two transmembrane domains and SUR1 has 17 - the channel itself is an octamer containing 4 molecules of each of these subunits. Control of channel opening by ATP and ADP has turned out to be considerably more complicated than at first expected. SUR1 is a so-called ATP-binding cassette (ABC) protein and contains two nucleotide binding domains. However closure of the channel is actually effected by binding of ATP to Kir6.2. The nucleotide binding sites on SUR1 bind MgADP causing conformational change in SUR1 which is transmitted to Kir6.2 to cause channel opening. Although the 3-dimensional structure of Kir6.2 has not yet been solved, a molecular model of Kir6.2 has been constructed by Mark Sansom and Fran Ashcroft, in Oxford, based upon the known structure of the bacterial Kir channel KirBac1.1 and of the N- and C-terminal domains of Kir3.1. The molecule is divided into two main regions with a helix, the ‘slide helix, at the interface between them. The TM domain has 2 membrane-spanning α-helices (TM1 and TM2) per subunit; these are connected by loop segments and a short α-helix (the ‘pore helix’) that loops part way into the membrane. The selectivity filter lies within the loop that links the pore helix to TM2. The N- and C-terminal moieties of each subunit interact extensively to form the IC domain and contain the ATP-binding sites. There are four ATP-binding sites, one per subunit. The adenine ring lies within a mainly hydrophobic pocket. There are H-bonds between N6 atom of the adenine ring and residues E179 (side chain) and R301 (carbonyl oxygen). The α-phosphate makes electrostatic interaction with R201, the ß-phosphate with K185 and the γ-phosphate with R50 from a neighbouring subunit. The γ-phosphate is not buried within the molecule as in most ATP-binding sites but projects out into the external solution. This explains why it is free ATP rather than MgATP that causes channel closure. How does channel closure occur? Since each ATP is bound to 2 subunits and each monomer is connected to 2 other subunits, ATP binding to 1 subunit will influence the relative conformations of adjacent subunits, thus explaining why binding of a single ATP molecule is sufficient to close the pore. It has been shown recently that mutation in R201 leads to permanent neonatal diabetes. The mutation causes decreased sensitivity to ATP. Other mutations found in PND are found in the slide helix, distinct from the ATP-binding-site.
What about control of SUR1 by nucleotides? The topology of SUR1 is thought to be as follows:-
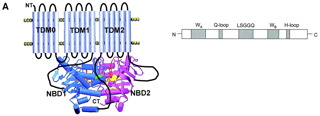
and indicates that NBDs contain several motifs involved in nucleotide binding and hydrolysis - the Walker-A and - B motifs, linked by a ‘signature sequence’ LLSGGQ and two shorter sequences containing conserved glutamine (Gln-loop) and histidine (His-loop) residues. Again the crystal structure of SUR1 is not known, but from the known structures of other ABC proteins such as CFTR a model for the ATP-binding site has been constructed. The X-ray structures so far determined for ABC proteins show that the NBDs form a dimer containing 2 nucleotide-binding pockets. These do not correspond to the NBDs encoded in the primary sequence; rather the ATP is sandwiched between the Walker-A of one NBD and the signature sequence of the other. Several mutations in SUR1 that lead to hyperinsulinism occur in the NBDs. For example E1506K may prevent interaction of Mg-nucleotides, and other mutations are close to the predicted ATP-binding pockets.
Thus the substrate site hypothesis as applied to glucose stimulation of Ca-entry into the ß-cell is now fully validated down to molecular details which are profoundly influencing our knowledge of ß-cell disease. However, glucose also causes Ca-mobilisation and this effect also depends on glucose metabolism. What are the mechanisms involved here? A complicating factor is that there are multiple routes possible for release of Ca from IC stores. The key points established from studies on various tissues are:-
1. Calcium is stored in the ER and also in lysosomes
2. Ca2+-transport into the ER occurs via the P-type ATPase SERCA
3. Ca2+-transport into lysosomal stores uses a H+-pump (a V-type ATPase) and a Ca2+-H+-exchanger
4. ER contains Ca2+-channels gated by IP3 or cADPR
5. The novel messenger NAADP mobilises Ca2+ from the lysosomal stores
cADPR and NAADP are synthesised by the same enzyme - cADPR cyclase
Interaction between Ca2+-pools via CICR can generate Ca2+-oscillations
In my most recent studies before retirement, in collaboration with Roser Masgrau and Antony Galione in Oxford we investigated these phenomena in the ß-cell. We found convincing evidence that NAADP indeed plays a key role in control of ß-cell Ca2+. The evidence is summarised as follows:-
1. MIN6 ß-cells contain specific NAADP receptors . By using radiolabelled NAADP we showed that NAADP bound to both high and low affinity sites and the binding was not displaced by IP3 or cADPR
2. NAADP is able to elicit an increase in ß-cell Ca2+. In these experiments cells were microinjected with a Ca-indicator and varying concentrations of caged NAADP and then exposed to UV.
3. Blocking NAADP receptors reduced glucose-stimulated insulin release.
4. Glucose increases NAADP concentration in ß-cells.
These data support the view that NAADP is an authentic second messenger in the ß-cell.
Where does NAADP act? To establish the sites of Ca-release in response to agonists we used inhibitors :- thapsigargin which depletes the ER Ca-pool by uptake of Ca2+ by SERCA; bafilomycin, which depletes the store of Ca2+ in lysosomes by blocking the vacuolar H+-ATPase that drives Ca-uptake.
Thapsigargin had no effect upon NAADP responses but eliminated IP3 responses. Conversely, bafilomycin A1 inhibited NAADP but had no effect upon IP3.Thus NAADP mobilises Ca from lysosomal stores whereas IP3 targets ER.
Do different extracellular signals, in particular glucose, use different second messengers? Comparison of the effects of bafilomycin and thapsigargin on Ca-responses in MIN6 cells induced by glucose, ACh and high K+ was striking. Bafilomycin abolished the Ca-signals induced by glucose but was without effect on those induced by ACh or high K+. The pattern of responses to thapsigargin was quite distinct. Thapsigargin had no effect on glucose-induced Ca-responses but abolished the response to ACh, and slightly potentiated the response to high K+. Thus it seems that the major Ca-store mobilised in response to glucose is an acidic store distinct from the endoplasmic reticulum. By recruiting NAADP, glucose selects a novel Ca-store with distinct properties, distribution and ramifications from the ER which is primarily the target for IP3 (and possibly cADPR).
The validation and amplification of the substrate-site hypothesis and the mechanisms underlying it have been the main theme of my work. However along the way I have been interested in a number of related topics. Some of the results are as follows.
I demonstrated that the substrate-site hypothesis was also applicable to the stimulatory effect of glucose on insulin biosynthesis and showed that the mechanism involved enhanced transcription of the insulin gene. I also initiated biochemical measurements on human pancreatic islets in 1971; described regulation and specificity of glucose stimulation of proinsulin gene expression in human islets in 1967 ; and discovered uncontrolled release of insulin from islets of a patient with nesidioblastosis (now known as persistent hyperinsulinaemia and hyperglycaemia of infants, PHHI).
Mary Sugden in my group showed that pancreatic islets contain high concentrations of calmodulin of functional importance for secretion; and we obtained evidence that Ca2+/ calmodulin-dependent protein kinase II (CaMKII) is an important target for calcium in the ß-cell I was one of the first to identify protein kinase C (PKC) in an endocrine gland and showed that multiple isoforms are present in the ß -cell and that PKC is functionally relevant to insulin secretion]. In collaboration with Graham Hardie, University of Dundee, I provided evidence that AMP-dependent protein kinase has a role in control of ß -cell function. These studies revealed that a major mechanism by which protein phosphorylation affects insulin secretion is through increased sensitivity to calcium.
Rebecca Ashfield in my group first cloned the promoter regions for Kir6.2 and SUR1 genes which has permitted study of the regulation of their expression. This is important given the highly selective tissue distribution of the proteins and the possibility for altered regulation of gene expression in Type II diabetes and/or PHHI. We have also been able to express functioning KATP channels in the baculovirus expression system. The production in this system of large quantities of purified KATP channel protein subunits has allowed my colleague Michael Mikhailov (now working in Fran Ashcroft's group) to make significant recent advances towards determining the 3-dimensional structure of the KATP channel.
ß -Cell Ca-ATPases: Following stimulation, ß -cells extrude Ca2+ from the cytosol by means of active Ca2+ pumps in the plasma membrane (PMCA) and endoplasmic reticulum (SERCA). These pumps are heterogeneous with both separate genes and differential splicing contributing to diversity. Using RT-PCR and immunological methods the PMCA and SERCA isoforms expressed by the ß -cell have been defined in collaborative studies with Dr Anikó Váradi. This work provided evidence that altered expression of the sarco(endo)plasmic reticulum Ca-pump, SERCA3, may contribute to the defective insulin secretion seen in an animal model of Type II diabetes .
CaM kinase II: my early studies implicated CaM kinase II in the control of insulin secretion. Since then we discovered a new ß -cell isoform of CaM kinase II, ß3, and cloned it from a rat ß -cell cDNA library. The ß -cell CaM kinase II ß3 contains an insertion of a unique duplicated proline-rich domain. The existence of the proline-rich domains as distinct exons in the human CaMKII ß gene has been confirmed. We showed that human islets of Langerhans also express CaM kinase II γ and δ isoforms and a novel truncated variant suggested to arise via trans-splicing.
Protein kinase C: Protein kinase C (PKC) is able to exert a number of effects on the ß -cell including a negative effect on ß -cell increases in [Ca2+] despite potentiation of insulin release. Using RT-PCR and Western and Northern blotting techniques Ya-Min Tian in my group established that the ß-cell expresses multiple isoforms of all the known classes of PKC - classical, novel and atypical - and obtained evidence that the response to cholinergic agonists may involve specifically α and ε isoforms.
Glutamate receptors: In collaboration with Dr E Molnár, University of Bristol, we demonstrated that ß-cells express ionotropic glutamate receptors and provided evidence for their functional role. We further showed that the ß -cell also expresses both metabotropic glutamate and GABA(B) receptors which contribute to the modulation of glucose-stimulated insulin secretion.
ß-Cell genes and Type II diabetes
A major aim of recent studies was to define precisely which isoforms and splice variants of proteins relevant to ß-cell function are expressed by the ß-cell. This has permitted a programme of screening for polymorphisms in order to identify mutations which may be linked to Type II diabetes. The main findings were as follows:-
SERCA genes: Screening of the entire coding and promoter regions of the human SERC3 gene by Dr Anikó Váradi found the presence of four rare missense mutations in exons 4, 14 and 15 in white Caucasian Type II diabetic subjects but not in control non-diabetics. The mutations affect residues that could cause impairment of Ca2+ uptake into the endoplasmic reticulum via SERCA3 and result in altered ß -cell Ca2+homeostasis. The patients with exon 4 or 14 mutations had a phenotype of normal body weight with marked hyperglycaemia and ß cell dysfunction. This is a rare phenotype found in only 6% of the Type II diabetics recruited for the UK Prospecive Diabetes Study. The data suggest that in white Caucasians the SERCA3 locus contributes to the genetic susceptibility to Type II diabetes
CaMKII genes: In collaboration with Dr Anna Gloyn we determined the structure of the human CaMKII gamma gene. The gene was localised to chromosome 10q22and shown to contain 23 exons. Screening of the 23 exons including the exon-intron boundaries for variants in subjects with Type II diabetes identified 2 variants (an exon 2 silent variant K49K and an intronic variant (exon 14). A small case-control study showed no association of the CaMKII gamma exon 2 variant with Type II diabetes; however the numbers of subjects studied was small and larger numbers of subjects need to be screened to investigate the role of this gene fully. The variants detected have allele frequencies of >10% which makes them suitable for family-based association studies to investigate the role of these genes in susceptibility to Type II diabetes.
KATP channel genes: Two variants were found in the human SUR1 promoter (a three base insertion (caa) at -522 bp and a single base substitution at -679 bp (c g)) and shown not to be associated with diabetes not were they in a sequence consensus region for transcription factors. Analysis of a variant in the Kir6.2 gene (E23K) showed that the KK homozygosity was more frequent in Type II diabetics than in control subjects. Anna Gloyn has gone on to make important discoveries on the role of KATP channel mutations in disorders of insulin secretion
Conclusions
Study of the mechanisms controlling insulin biosynthesis and release from the ß-cell has been a challenging and exciting research area. It has been for me a pleasure to have been involved with the development of the field which has seen ideas become reality and move towards established mechanisms in which molecular details are becoming apparent. And the recent explosion in knowledge of the relevance of the findings to human disease has been particularly satisfying.